Among the genes involved in neuronal differentiation, only a few have been studied in relation to Folinic acid calcium salt pentahydrate regulatory control by nuclear FGFR1, Nurs and RA receptors. Nuclear FGFR1 increases the expression of th, neurofilament, neuronal enolase and fgf-2 and chromatin immunoprecipitation experiments showed nuclear FGFR1, together with CBP and other DNA binding proteins, associates within the LOUREIRIN-B promoters of the th and fgf-2 genes. Yeast two-hybrid and coimmunopreciptation assays revealed that the FGFR1 tyrosine kinase domain binds directly to RSK1 Nterminal kinase. RSK1 binding promotes FGFR1 release from pre-Golgi to cytosol, increases the mobile population cytosolic of FGFR1 and facilitates nuclear accumulation of FGFR1. In the cell nucleus interaction of FGFR1 with RSK1 restricts the FGFR1 intra nuclear mobility and promotes RSK1 activation of CREB. In addition our recent studies showed that FGFR1 forms nuclear complexes with both the Nurr1 and Nur77 proteins. Given that RSK and Nur77 are fundamentally involved in nuclear signaling through both NGF and INFS, the possibility that NGF may utilize the INFS mechanism for neurodevelopmental and gene-activating functions has now been examined. We report that NGF promotes FGFR1 cytoplasmic-nuclear trafficking, in part, by inhibiting FGFR1 nuclear export. Furthermore, nuclear FGFR1 is essential for NGF-induced differentiation and transcriptional programming ofPC12 cells.FGFR1 binds to Nurtargeted regions of NGF-activated genes and augments NGF activation of ligand-independent function of Nur77/Nurr1. The present study provides a new perspective on the diverse actions of NGF which requires the neurodevelopmental INFS mechanism. Earlier studies from several laboratories 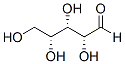 have provided in depth characterization of NGF-induced PC12 neuronal-like differentiation i.e., an outgrowth of neurites with growth cone-like endings accompanied by an up-regulation of the neurotransmitter biosynthetic enzyme, TH, neuronal b-III Tubulin, MAP-2, Neurofilament L, NMDAR1 protein and nicotinic acetylcholine receptor currents. Other studies have also indicated that NGF can evoke neuron-specific voltagedependent K+ and Na+ currents. In the present work we find that NGF-induced nuclear accumulation of FGFR1 is accompanied by exit from the cell cycle, an acquisition of neuronal morphology and the activation of th-Luc and other neuronal genes. The outgrowth of PC12 neurites was analyzed by measuring the length of neuritic processes using an established assay in cells co-transfected with plasmid expressing marker EGFP protein. Treatment of PC12 cells with NGF produced typical neurite outgrowth. In a loss of function experiment we cotransfected PC12 cells with dominant negative mutants of FGFR1, which lack the tyrosine kinase domain, form non-functional dimers with the endogenous receptor and compete with wild type FGFR1 for its nuclear targets. FGFR1 localizes to cytoplasmic membranes and cell nuclei. FGFR1, in which the signal peptide is replaced with a NLS, functions exclusively in the nucleus. Cells co-transfected with a control vector display short processes, however, when treated with NGF the processes elongate. The dominant negative receptors have no significant effect on neurite length in nonstimulated PC12 cells compared to controls. In contrast, cells transfected with FGFR1 or FGFR1 fail to extend neurites in response to NGF. In a gain of function experiment, PC12 co-transfected with full length constitutiveFGFR1, which contains a functional tyrosine kinase domain, display a marked 3-foldelongation of neurites indistinguishable from that induced by NGF. The effects of FGFR1 on neurite outgrowth are summarized in Fig. 3B. Additional experiments show that NGF increases the number of PC12 cells with elongated process while reducing the cell number with short processes. This effect is diminished by dominant negative FGFR1.
have provided in depth characterization of NGF-induced PC12 neuronal-like differentiation i.e., an outgrowth of neurites with growth cone-like endings accompanied by an up-regulation of the neurotransmitter biosynthetic enzyme, TH, neuronal b-III Tubulin, MAP-2, Neurofilament L, NMDAR1 protein and nicotinic acetylcholine receptor currents. Other studies have also indicated that NGF can evoke neuron-specific voltagedependent K+ and Na+ currents. In the present work we find that NGF-induced nuclear accumulation of FGFR1 is accompanied by exit from the cell cycle, an acquisition of neuronal morphology and the activation of th-Luc and other neuronal genes. The outgrowth of PC12 neurites was analyzed by measuring the length of neuritic processes using an established assay in cells co-transfected with plasmid expressing marker EGFP protein. Treatment of PC12 cells with NGF produced typical neurite outgrowth. In a loss of function experiment we cotransfected PC12 cells with dominant negative mutants of FGFR1, which lack the tyrosine kinase domain, form non-functional dimers with the endogenous receptor and compete with wild type FGFR1 for its nuclear targets. FGFR1 localizes to cytoplasmic membranes and cell nuclei. FGFR1, in which the signal peptide is replaced with a NLS, functions exclusively in the nucleus. Cells co-transfected with a control vector display short processes, however, when treated with NGF the processes elongate. The dominant negative receptors have no significant effect on neurite length in nonstimulated PC12 cells compared to controls. In contrast, cells transfected with FGFR1 or FGFR1 fail to extend neurites in response to NGF. In a gain of function experiment, PC12 co-transfected with full length constitutiveFGFR1, which contains a functional tyrosine kinase domain, display a marked 3-foldelongation of neurites indistinguishable from that induced by NGF. The effects of FGFR1 on neurite outgrowth are summarized in Fig. 3B. Additional experiments show that NGF increases the number of PC12 cells with elongated process while reducing the cell number with short processes. This effect is diminished by dominant negative FGFR1.