Lastly, our work underscores the role of negative regulators of receptor RTKs in cellular utilization of these receptors and should be taken into consideration for drug response evaluation of any molecular targeted therapies to other RTKs. Epigenetic changes are increasingly recognised as a major characteristic of many human diseases. Almost all CpG dinucleotides are methylated, except those located in CpG islands, which lack DNA methylation setting them apart from bulk genomic DNA. Aberrant methylation of CGIs in or near the promoter region of tumour suppressor genes represents one of the most consistent hallmarks of human cancers and these TSGs are often silenced in haematopoietic malignancies. Thus, CGI methylation represents an ideal candidate for diagnostic and prognostic cancer markers. Myelodysplastic syndromes comprise a heterogeneous group of bone marrow disorders affecting mainly elderly patients. A number of gene mutations and cytogenetic changes have been implicated in the pathogenesis of MDS, including mutations of RAS, TP53 and RUNX1, and more recently ASXL1, c-CBL, DNMT3A, IDH1/2, TET2, and EZH2. Nevertheless, these genetic abnormalities do not fully explain the pathogenesis of MDS because they are also commonly found in other myeloid malignancies and roughly 20% of MDS patients have no known genetic mutation. On the other hand, hypermethylation of specific genes, such as p15, E-cadherin, ER, MYOD1, and HIC1, have been noted, and whole genome studies have revealed that MDS patients contain aberrant DNA methylation in thousands of genes compared to normal haematopoietic progenitor cells. The process of cytosine methylation is reversible and may be altered by biochemical and biological manipulation, making it an attractive target for therapeutic intervention. Epigenetic therapy with hypomethylating drugs is now the standard of care for MDS. Two prominent examples are the cytosine analogs 5-azacytidine and 2′-deoxy-5-azacytidine. These are potent inhibitors of DNA methyltransferases and have been approved for MDS treatment. Recent efforts have focused on lowering the dosage of azacytidine and decitabine to reduce toxicity. However, the effect of low-dose treatment on the MDS methylome is still unclear. In this report, we have determined concentrations of AZA and DAC that allow prolonged treatment in a leukemic cell model, and have determined how this affects global CGI methylation using a microarray approach. Our results show that the methylome was selectively demethylated by lowdose treatments and that gene-body CGIs were more resistant to this process. We also provide evidence that prolonged lowdose AZA and DAC treatment is sustainably effective in modifying the epigenome. We next examined the correlation between expression and methylation levels. We performed transcriptome analyses for both mock and drug AB1010 VEGFR/PDGFR inhibitor treated SKM-1 cells. The level of methylation in individual islands was summarised by the mean log2-ratios, and these were plotted against expression levels. Since individual genes can overlap multiple CGIs we divided the CGIs into classes depending on their overlap with gene features as described above and made separate plots for each class. In the control cells, a clear anticorrelation between gene expression and methylation was observed for CGIs overlapping promoter elements. This correlation was stronger for promoter CGIs with low CG content, which may be due to the general paucity of highly methylated high CG density CGIs. The data also suggested that relationships between expression levels and DNA methylation exist at non-promoter CGIs. However, these relationships were not as robust with Nutlin-3 observations depending on the summary statistics used, and apparently restricted to subsets of islands within each 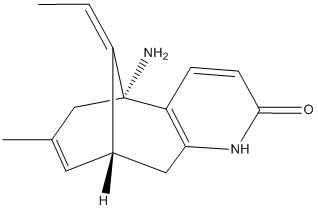 class rather than generally true for the full set of islands. Interestingly, this anti-correlation was lost or markedly reduced in AZA and DAC treated cells. However, expression levels within AZA and DAC treated cells were still anti-correlated against promoter methylation levels in control cells. This strongly suggests that promoter CGI demethylation was not generally sufficient to modify expression patterns, and emphasizes the roles of other means of maintaining cell state. Although a correlation between CGI demethylation and upregulation of gene expression was not generally observed, we identified a small number of genes where expression appeared to change following demethylation.
class rather than generally true for the full set of islands. Interestingly, this anti-correlation was lost or markedly reduced in AZA and DAC treated cells. However, expression levels within AZA and DAC treated cells were still anti-correlated against promoter methylation levels in control cells. This strongly suggests that promoter CGI demethylation was not generally sufficient to modify expression patterns, and emphasizes the roles of other means of maintaining cell state. Although a correlation between CGI demethylation and upregulation of gene expression was not generally observed, we identified a small number of genes where expression appeared to change following demethylation.
Month: July 2019
Prolonged ISA27 exposure caused a further decline in viable cells due to a G2-phase cell cycle block and apoptosis
Effect of the two drugs was represented by the straight line connecting the two points. If the experimentally determined data points and their confidence interval fall on this line, the drug effects are additive. If the points lie below this line, there is superadditivity, and if the points lie above this line, there is subadditivity. To Nutlin-3 determine whether the interaction between the two drugs was synergistic, additive or antagonistic, the theoretical additive IC50,add was estimated from the dose-response curves of each drug administered individually, considering that the observed effect with the combination was the sum of the individual effects of each component. The theoretical IC50,add value was then compared with the experimental IC50,mix to determine whether there was a statistically significant difference. The interaction index, denoted by c, is an assessment of the degree of synergism or antagonism. The index is defined by the isobolar relationship as follows : c=a/A+b/B where A and B are the doses of drug A and B that give the specified effect, and are the combination doses that produce the same effect. The quantities in the equation are obtained from the dose-response curves of drugs A, B, and their combinations. If c=1, the interaction is additive; if c,1, the interaction is super-additive ; and if c.1, the interaction is sub-additive. The direct and specific activation of the p53 pathway without inducing collateral DNA damage offers a tantalising solution to the shortcomings of current 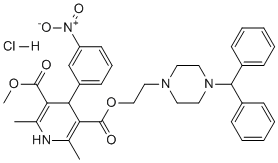 therapeutic regimens and appears to be a reasonable approach for GBM therapy in view of the infrequent occurrence of p53 gene mutations. The cumulative evidence of aberrantly increased activity of the primary p53 inhibitor MDM2 in GBM prompted us to examine the effects of targeted inhibition of the MDM2-p53 interaction by the spiro-oxindole analogue ISA27, a recently described smallmolecule inhibitor of MDM2. Little is known about the effects of MDM2 inhibitors on the in vitro growth of GBM cells. Recently, Nutlin-3, the first potent small-molecule inhibitor of MDM2, was reported to be effective at inhibiting GBM cell growth in vitro, suggesting the validity of this experimental approach for the treatment of GBM. In this study, we investigated whether ISA27 affected the growth of GBM cells and explored the intracellular events following ISA27 treatment. The U87MG and U343MG cell lines overexpress MDM2 and maintain wild-type p53 and were chosen as a cell culture model of human GBM. In these cell lines, the primary mechanism of p53 inactivation is the high nuclear MDM2 levels caused by a lack of PTEN, a tumor suppressor protein that normally counteracts MDM2 translocation into the nucleus. The lack of PTEN makes these cell lines a suitable representative model of GBM, as the loss of the PTEN gene locus has been found in up to 80% of GBM cases. This is the first report to demonstrate that ISA27 is a potent inhibitor of GBM cell growth. Previous studies have shown that ISA27 activates p53, resulting in growth inhibition in HEK-293 transformed human embryonic kidney, M14 human melanoma and U937 human monocyte lymphoma cell lines. Our results demonstrate that ISA27 blocks the cell cycle and triggers an apoptotic cell death program in the GBM cells, responses that are similar to those obtained in human M14 melanoma cells. We observed a dose-dependent PLX-4720 918505-84-7 antiproliferative effect of ISA27 in U87MG and U343 cells following short-term treatments with increasing ISA27 concentrations. The specificity of the antiproliferative effects was demonstrated by the accumulation of the p53 protein due to the decreased interaction between p53 and MDM2 and the restoration of p53 function in GBM cells after ISA27 treatment. The reactivation of p53 was suggested by the transcriptional activation of the primary target genes of p53, MDM2 and CDKN1A. The CDKN1A gene encodes the cyclin-dependent kinase inhibitor p21, an essential mediator of p53-induced cell cycle arrest. Exponential growth of U87MG cells was significantly inhibited following long-term ISA27 treatment. The number of viable cells was substantially reduced after 1 day of ISA27 treatment, and this reduction reached almost total growth inhibition after 5 days. Kinetic analyses of the ISA27-induced intracellular effects showed that the decrease in viable cells was mainly due to a G1-phase cell cycle block and cellular senescence during the initial phase of ISA27 treatment. However, apoptotic parameters, such as the dissipation of mitochondrial membrane potential, began to appear.
therapeutic regimens and appears to be a reasonable approach for GBM therapy in view of the infrequent occurrence of p53 gene mutations. The cumulative evidence of aberrantly increased activity of the primary p53 inhibitor MDM2 in GBM prompted us to examine the effects of targeted inhibition of the MDM2-p53 interaction by the spiro-oxindole analogue ISA27, a recently described smallmolecule inhibitor of MDM2. Little is known about the effects of MDM2 inhibitors on the in vitro growth of GBM cells. Recently, Nutlin-3, the first potent small-molecule inhibitor of MDM2, was reported to be effective at inhibiting GBM cell growth in vitro, suggesting the validity of this experimental approach for the treatment of GBM. In this study, we investigated whether ISA27 affected the growth of GBM cells and explored the intracellular events following ISA27 treatment. The U87MG and U343MG cell lines overexpress MDM2 and maintain wild-type p53 and were chosen as a cell culture model of human GBM. In these cell lines, the primary mechanism of p53 inactivation is the high nuclear MDM2 levels caused by a lack of PTEN, a tumor suppressor protein that normally counteracts MDM2 translocation into the nucleus. The lack of PTEN makes these cell lines a suitable representative model of GBM, as the loss of the PTEN gene locus has been found in up to 80% of GBM cases. This is the first report to demonstrate that ISA27 is a potent inhibitor of GBM cell growth. Previous studies have shown that ISA27 activates p53, resulting in growth inhibition in HEK-293 transformed human embryonic kidney, M14 human melanoma and U937 human monocyte lymphoma cell lines. Our results demonstrate that ISA27 blocks the cell cycle and triggers an apoptotic cell death program in the GBM cells, responses that are similar to those obtained in human M14 melanoma cells. We observed a dose-dependent PLX-4720 918505-84-7 antiproliferative effect of ISA27 in U87MG and U343 cells following short-term treatments with increasing ISA27 concentrations. The specificity of the antiproliferative effects was demonstrated by the accumulation of the p53 protein due to the decreased interaction between p53 and MDM2 and the restoration of p53 function in GBM cells after ISA27 treatment. The reactivation of p53 was suggested by the transcriptional activation of the primary target genes of p53, MDM2 and CDKN1A. The CDKN1A gene encodes the cyclin-dependent kinase inhibitor p21, an essential mediator of p53-induced cell cycle arrest. Exponential growth of U87MG cells was significantly inhibited following long-term ISA27 treatment. The number of viable cells was substantially reduced after 1 day of ISA27 treatment, and this reduction reached almost total growth inhibition after 5 days. Kinetic analyses of the ISA27-induced intracellular effects showed that the decrease in viable cells was mainly due to a G1-phase cell cycle block and cellular senescence during the initial phase of ISA27 treatment. However, apoptotic parameters, such as the dissipation of mitochondrial membrane potential, began to appear.
In the NA gene other than H274Y and is therefore considered as a natural variant of previous strains
As the influenza virus life cycle critically depends on a balance between available receptor sitesand receptor binding, the new variant may have emerged by selection of a compensatory co-mutation in the hemagglutinin gene to acquire full virulence. Polysaccharides and in particular carrageenans were found to be potent antiviral agents against certain viruses. The antiviral effects of carrageenans were of limited practical importance so far, most likely because carrageenans are high-molecular weight components making it unlikely that they pass the different barriers of the body or even the cell membrane. However, these characteristics do not rule out local applications. A recent study with Carraguard, a carrageenan-based compound developed by the Population Council, did not show efficacy in prevention of vaginal transmission of HIV. The authors conclude that low acceptance of gel use could have compromised the potential to detect a significant protective effect of Carraguard. In contrast to influenza viruses, HIV causes a persistent systemic infection that is usually not cleared by the immune system of the organism. Therefore, an incomplete protective effect at the entry site of the virus might lead to full blown HIV infection that is inaccessible to treatment with an antiviral polymer. The results of our animal experiments allow the speculation that treatment with iota-carrageenan reduced the spreading of influenza virus in surface epithelia of infected animals and thereby provided sufficient benefit for animals to promote survival. In conclusion, our results suggest that iota-carrageenan is safe and effective in treating influenza infection in an animal model. Moreover, given that a iota-carrageenan-containing nasal spray is already marketed in Europe and has successfully been tested in an exploratory trial for treating common cold in humans, iotacarrageenan is also a promising antiviral candidate for the prophylaxis and treatment of influenza virus infections and should be tested for prevention and treatment of influenza A in clinical DAPT inquirer trials in humans. Besides their critical role in intra- and intercellular “waste management”, proteases are currently accepted as important signaling molecules involved in numerous biological and pathological functions. These include metabolism, tissue remodeling, apoptosis, cell proliferation and migration. Thus, protease signaling needs to be strictly regulated, and the deregulation of protease activity may contribute to various pathologies, including neoplastic diseases. The human Threonine Aspartase 1/Taspase1 gene encodes a protein of 420 amino acids, representing the proenzyme of the protease. In contrast to the other exclusively cis-active type 2 Asparaginases, only Taspase1 is also able to cleave other substrates in trans. Therefore, Taspase1 represents a distinct class of proteolytic enzymes. Taspase1 mediates LDK378 ALK inhibitor cleavage of proteins by recognizing a conserved peptide motif with an aspartate at the P1 position. The N-terminal threonineis generated by autoproteolysis of the Taspase1 proenzymeinto the two subunits a and b, which appear to assemble into an asymmetric 28 kDa/22 kDa a2/b2-heterotetramer, the active protease. The discovery of Taspase1 founded a new class of endopeptidases that utilize the N-terminal threonine of its mature b-subunit as the active site. Mutation of this catalytic nucleophile, Thr234, abolishes Taspase1��s proteolytic activity. Taspase1 was first identified as the ![]() protease responsible for cleavage of the Mixed Lineage Leukemiaprotein at conservedsites. Proteolytic cleavage of MLL is considered to stabilize the MLL proteinas a crucial event for proper Hox gene expression and normal cell cycle.
protease responsible for cleavage of the Mixed Lineage Leukemiaprotein at conservedsites. Proteolytic cleavage of MLL is considered to stabilize the MLL proteinas a crucial event for proper Hox gene expression and normal cell cycle.
When aliquots of this culture were transferred onto YPD plates discovered that chemical treatment
The growth rates of BY4741 and AD1-9 in the presence of all compounds tested are depicted in Figure S1. As expected, growth of the drug-efflux pump deficient strain was more often and more strongly inhibited than that of the wild-type strain. Altogether, 231 compounds inhibited growth of BY4741 and/or AD1-9. To identify meiosis-specific inhibitors, all drugs in the NCC were subsequently interrogated with the two sporulation assays. For these experiments we used the efficiently-sporulating SK1 strain background, which was not deficient in any of the drugefflux pumps. Similar to the growth data, we calculated sensitivity scores for every compound. This score indicates how strongly a compound inhibited sporulation in the assay compared to the “no drug” controls. To minimize false positives and prioritize secondary experiments, a highly stringent cutoff was applied. A scatterplot depicts the sensitivity scores ABT-263 determined this wayand a Venn diagram summarizes the overlap of compounds identified in the three different screening approaches. In summary, 200 drugs had no effect in any of the assays; 231 inhibited growth and 64 inhibited sporulation. 49 drugs inhibited vegetative growth and sporulation. All sensitivity scores are listed in Table S1. 15 compounds were found to specifically inhibit spore formation. We re-tested these chemicals in sporulating cultures and determined the percentage of spores after 48 hours. Note that these experiments were performed in small liquid cultures, which are sub-optimally aerated, and thus only intermediate sporulation efficiencies were observed. Nevertheless, 10 of the 15 compounds completely abolished spore formation and 2 chemicalshad moderate inhibitory effects. The remaining 3 compounds were indistinguishable from the control and were not further analyzed. Taken together, the Talazoparib 1207456-01-6 confirmation rate was 80%. Five of the twelve compoundswere identified in the sporulation assay only. Another set of 5 drugswas identified in the recombination assay only. The remaining 2 compoundswere identified in both assays. We noticed a similarity in molecular structures among the inhibitory compounds: all contained a hydrophobic ring system and a basic nitrogen-containing group, which are the attributes of a class of compounds called cationic amphiphilic drugs. The primary targets of these drugs in man are monoamine receptors, and many are widely used anti-depressants and antipsychotics. These drugs are not only known to interact with their protein targets, but also with phospholipid membranes. Yeast lack proteins with sequence similarity to monoamine receptors, therefore, these drugs likely repress sporulation by inhibiting alternative proteins or cellular components. We chose tripelennamineas a representative for this class of compounds, and studied its effect on yeast sporulation in more detail. We first determined sporulation efficiency in the presence of TA in largeliquid cultures. As expected the “no drug” control yielded,95% mature spores within the first 24 hours, and ammonium sulfate strongly reduced spore formation to only about 10% after 3 days. In contrast, TA completely abolished sporulation at a concentration of 100 mM. Even after 72 hours no spores were detected in the TA-treated culture. Upon visual inspection of sporulating cultures by microscopy, marked morphological differences were observed in these three conditions. In the “no 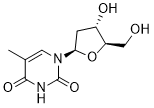 drug” control, most cells had formed an ascus with spores, whereas the ammonium sulfate-treated cells had round and inflated shapes. In contrast, cells that were sporulated in the presence of TA had accumulated small granular bodies of unknown nature, but were devoid of spores.
drug” control, most cells had formed an ascus with spores, whereas the ammonium sulfate-treated cells had round and inflated shapes. In contrast, cells that were sporulated in the presence of TA had accumulated small granular bodies of unknown nature, but were devoid of spores.
It explains the structure activity relationship of the IWRs and will be important for further
For example, the critical His1201 from the D-loop of TNKS1is not conserved in other PARP proteins; the a3 helix N-terminal to the D-loop is slightly shorter in tankyrases due to the insertion of a prolineand deletion of two amino acids, resulting in a narrower induced pocket. Therefore, one is likely to achieve broader selectivity over PARP family members with compounds that bind to the induced pocket. For example, the selectivity of XAV939 for TNKS1 over PARP2 is only 10 fold whereas the selectivity of 2 is greater than 143 fold. The TNKS1/2 complex structure and molecular modeling analysis suggest a number of distinct routes to further optimize tankyrase inhibitors that bind to the induced pocket. Preliminary metabolic stability studies indicated enzymatic cleavage of the amide bond to be the primary clearance mechanism for IWRs. It is clear from our crystal structure that the amide quinoline ![]() of 2 can be replaced by other more stable moieties that maintain the same hydrogen bonding and stacking interactions. Modifications of the central phenyl group may also generate compounds with more favorable binding geometries. Quantum mechanical calculations suggest that the,60u dihedral between the phenyl and amide Fingolimod supply observed in the crystal structure of 2 results in an intrinsic reduction of potency by approximately 25-fold. The pyrrolidine dione group also does not appear optimal for tankyrase binding. One of the two carbonyl oxygens is not involved in hydrogen bonding or any other interaction with the protein and thus could be replaced. In addition, it is also conceivable that the norbornyl group does not interact optimally with the Tyr1213, Tyr1224, and Ile1228 of TNKS1. Furthermore, since the induced pocket is adjacent to the nicotinamide pocket which is unoccupied and unhindered, it may be possible to extend the induced pocket binding tankyrase inhibitors such as 2 into the nicotinamide pocket to gain additional interactions, resulting in even greater potency while maintaining good selectivity due to the specificity of the induced pocket. IWR compounds may have activity for proteins other than PARP family members; thus, minimizing potential side effects from the off-target interactions is essential for further development of tankyrase inhibitors derived from IWRs. Future studies such as chemical proteomics screens need to be carried out to identify potential unintended INCB28060 targets of these inhibitors. We note that induced pockets have been observed for other enzymes such as protein kinases. An allosteric binding pocket was reported for a diaryl urea class of highly potent and selective inhibitors against human p38 MAP kinase and the formation of this pocket requires a large conformation change. Improving interactions in this allosteric pocket and establishing additional interactions in the adjacent ATP pocket enhanced the affinity of the inhibitors by 12,000 fold. Imatinib, developed to treat chronic myelogenous leukemiaand gastrointestinal stromal tumor, binds to similar sites in the human Abl and Kit kinases and shows excellent efficacy and specificity for Abl and Kit. Interestingly, imatinib was found to inhibit stronglya non-kinase target, the oxidoreductase NQO2, from a screen carried out to identify off-target proteins. Vemurafenib, developed for the treatment of metastatic melanoma caused by the BRAFV600E mutation, also binds to an induced pocket created by an outward shift of the aC helix. In summary, the present structure reveals a novel binding mode for tankyrase inhibitors and, in conjunction with molecular modeling analysis, provides insights into the molecular basis for the key interactions between IWRs and tankyrases.
of 2 can be replaced by other more stable moieties that maintain the same hydrogen bonding and stacking interactions. Modifications of the central phenyl group may also generate compounds with more favorable binding geometries. Quantum mechanical calculations suggest that the,60u dihedral between the phenyl and amide Fingolimod supply observed in the crystal structure of 2 results in an intrinsic reduction of potency by approximately 25-fold. The pyrrolidine dione group also does not appear optimal for tankyrase binding. One of the two carbonyl oxygens is not involved in hydrogen bonding or any other interaction with the protein and thus could be replaced. In addition, it is also conceivable that the norbornyl group does not interact optimally with the Tyr1213, Tyr1224, and Ile1228 of TNKS1. Furthermore, since the induced pocket is adjacent to the nicotinamide pocket which is unoccupied and unhindered, it may be possible to extend the induced pocket binding tankyrase inhibitors such as 2 into the nicotinamide pocket to gain additional interactions, resulting in even greater potency while maintaining good selectivity due to the specificity of the induced pocket. IWR compounds may have activity for proteins other than PARP family members; thus, minimizing potential side effects from the off-target interactions is essential for further development of tankyrase inhibitors derived from IWRs. Future studies such as chemical proteomics screens need to be carried out to identify potential unintended INCB28060 targets of these inhibitors. We note that induced pockets have been observed for other enzymes such as protein kinases. An allosteric binding pocket was reported for a diaryl urea class of highly potent and selective inhibitors against human p38 MAP kinase and the formation of this pocket requires a large conformation change. Improving interactions in this allosteric pocket and establishing additional interactions in the adjacent ATP pocket enhanced the affinity of the inhibitors by 12,000 fold. Imatinib, developed to treat chronic myelogenous leukemiaand gastrointestinal stromal tumor, binds to similar sites in the human Abl and Kit kinases and shows excellent efficacy and specificity for Abl and Kit. Interestingly, imatinib was found to inhibit stronglya non-kinase target, the oxidoreductase NQO2, from a screen carried out to identify off-target proteins. Vemurafenib, developed for the treatment of metastatic melanoma caused by the BRAFV600E mutation, also binds to an induced pocket created by an outward shift of the aC helix. In summary, the present structure reveals a novel binding mode for tankyrase inhibitors and, in conjunction with molecular modeling analysis, provides insights into the molecular basis for the key interactions between IWRs and tankyrases.