The potential to interfere with these processes. Early studies demonstrated that nitrogen-containing compounds, such as amino acids and ammonium ions prevent yeast cells from sporulating. Other work described the effects of chemicals that induce aneuploidy  in yeast undergoing meiosis. Anti-neoplastic drugs, such as adriamycin, mitomycin C, and bleomycin were shown to disrupt the second meiotic division leading to the generation of diploid spores. These drugs, however, are not only effective during sporulation, but also abolish vegetative growth. In this study we aimed to identify chemicals that inhibit meiotic development in yeast but do not interfere with vegetative growth. We profiled a library of 446 drugs from the NIH clinical collection with two sporulation assays, and generated sensitivity profiles of growing and sporulating cells for each of these chemicals. This approach identified 12 potent, sporulation-specific inhibitors, the majority of which are cationic amphiphilic drugs. We have studied the effects of one of these drugs, tripelennamine, on various meiotic landmarks and identified genes related to autophagy as hypersensitive to the drug using chemical genomic profiling. Previous analyses of meiotic mutants in yeast have shown that cells can omit certain stages of meiotic development and still produce mature meiotic products. For example spo11D mutants, that are unable to perform meiotic recombination, are still FG-4592 capable of producing mature asci. Thus, chemical compounds that for example inhibit Spo11 would not be identified with the fluorescence-based assay described above. To overcome this limitation a second screening approach was employed. This approach is based on a hetero-allelic reporter system that has been used by others to measure meiotic reciprocal recombination, crossover and non-crossover recombination, and recombination frequencies. A strain harboring the his4 mutant allelesis unable to grow in the absence of histidine. Upon meiotic recombination between the two alleles, one of the four meiotic products will receive a functional HIS4 allele, generating a histidine-prototrophic cell that is capable of growing in the absence of histidine. This event is facilitated by the presence of two recombination hot-spots located within the HIS4 openreading frame. The production of histidine-prototrophs can be monitored by transferring aliquots of sporulating cells to media lacking histidine. Compounds that inhibit entry into meiosis, premeiotic DNA replication or recombination will suppress recombination between the his4 alleles and will therefore suppress the generation of such prototrophs. To validate this reporter assay, a proof-of-concept experiment was performed in which different concentrations of ammonium sulfate were added to his4x/his4B harboring cells upon induction of meiosis. After 5 hours of sporulation, where most cells have ABT-199 1257044-40-8 undergone pre-meiotic DNA-synthesis and meiotic recombination but have not undergone the commitment and can therefore return to growth, aliquots of the cultures were plated onto agar plates lacking histidine. As expected, the quantity of histidineprototrophic cells increased with decreasing concentrations of ammonium sulfate in the media. Results from this assay correlated with those from the fluorescence-based assay: 2, 1, and 0.5 mM of ammonium sulfate suppressed colony formation; lower concentrations of ammonium sulfate did not interfere with meiotic recombination and hence colony growth. Note, that in addition to compounds that specifically inhibit meiotic recombination and/or spore formation, the two screening assays described here will also identify compounds that are cytotoxic in cells undergoing these processes.
in yeast undergoing meiosis. Anti-neoplastic drugs, such as adriamycin, mitomycin C, and bleomycin were shown to disrupt the second meiotic division leading to the generation of diploid spores. These drugs, however, are not only effective during sporulation, but also abolish vegetative growth. In this study we aimed to identify chemicals that inhibit meiotic development in yeast but do not interfere with vegetative growth. We profiled a library of 446 drugs from the NIH clinical collection with two sporulation assays, and generated sensitivity profiles of growing and sporulating cells for each of these chemicals. This approach identified 12 potent, sporulation-specific inhibitors, the majority of which are cationic amphiphilic drugs. We have studied the effects of one of these drugs, tripelennamine, on various meiotic landmarks and identified genes related to autophagy as hypersensitive to the drug using chemical genomic profiling. Previous analyses of meiotic mutants in yeast have shown that cells can omit certain stages of meiotic development and still produce mature meiotic products. For example spo11D mutants, that are unable to perform meiotic recombination, are still FG-4592 capable of producing mature asci. Thus, chemical compounds that for example inhibit Spo11 would not be identified with the fluorescence-based assay described above. To overcome this limitation a second screening approach was employed. This approach is based on a hetero-allelic reporter system that has been used by others to measure meiotic reciprocal recombination, crossover and non-crossover recombination, and recombination frequencies. A strain harboring the his4 mutant allelesis unable to grow in the absence of histidine. Upon meiotic recombination between the two alleles, one of the four meiotic products will receive a functional HIS4 allele, generating a histidine-prototrophic cell that is capable of growing in the absence of histidine. This event is facilitated by the presence of two recombination hot-spots located within the HIS4 openreading frame. The production of histidine-prototrophs can be monitored by transferring aliquots of sporulating cells to media lacking histidine. Compounds that inhibit entry into meiosis, premeiotic DNA replication or recombination will suppress recombination between the his4 alleles and will therefore suppress the generation of such prototrophs. To validate this reporter assay, a proof-of-concept experiment was performed in which different concentrations of ammonium sulfate were added to his4x/his4B harboring cells upon induction of meiosis. After 5 hours of sporulation, where most cells have ABT-199 1257044-40-8 undergone pre-meiotic DNA-synthesis and meiotic recombination but have not undergone the commitment and can therefore return to growth, aliquots of the cultures were plated onto agar plates lacking histidine. As expected, the quantity of histidineprototrophic cells increased with decreasing concentrations of ammonium sulfate in the media. Results from this assay correlated with those from the fluorescence-based assay: 2, 1, and 0.5 mM of ammonium sulfate suppressed colony formation; lower concentrations of ammonium sulfate did not interfere with meiotic recombination and hence colony growth. Note, that in addition to compounds that specifically inhibit meiotic recombination and/or spore formation, the two screening assays described here will also identify compounds that are cytotoxic in cells undergoing these processes.
Month: July 2019
This treatment strategy synergistically decreased viability and promoted G1 cell cycle arrest even in the cell lines
To understand the basis for cell sensitivity to temsirolimus as a single agent, we turned to an analysis of Akt activation, both at baseline and in response to treatment. We found that the baseline constitutive activation of Akt was predictive of cell sensitivity with the most resistant cells having very low basal S473 and T308 phosphorylation. After treatment with temsirolimus, a compensatory increase in Akt phosphorylation at both sites was detected in the most sensitive endometrial cancer cell lines tested, but the primarily resistant cellsdemonstrated no Akt phosphorylation at either site, and another resistant line, Hec50co, showed reduced phosphorylation. Thus, in contrast to sensitive cells, primarily resistant cells have low basal Akt phosphorylation and do not respond with compensatory hyper-phosphorylation after temsirolimus treatment. Of note, phospho-PDK1, the kinase responsible for Akt phosphorylation at T308, is low in several resistant cell lines such as Hec1A and KLE. This indicates primary cell resistance and a general lack of dependence on the Akt BMS-354825 signaling pathway for proliferation. On the other hand, the finding of compensatory activation of Akt in responsive cells is consistent with previous reports in the literature of rapalog-induced Akt phosphorylation in many cancer cell lines, human xenograft models, and patient tumors. Compensatory hyper-Akt phosphorylation suggests one potential mechanism whereby cells that are initially sensitive escape the growth inhibitory effects of the drug and become secondarily resistant. From these data, we distinguish between primary resistance with a lack of baseline Akt dependence versus acquired resistance demonstrated by hyper-Akt activation in response to temsirolimus. Temsirolimus is currently in phase II trials for advanced endometrial cancer and has shown some promise. However, lack of initial response to therapy as well as the development of acquired drug resistance continues to be problematic. To more fully understand the therapeutic potential of mTOR inhibition in endometrial cancer, we first examined the effect of temsirolimus alone on the viability of a panel of endometrial cancer cell lines. We sought to distinguish between cellular events which predict for primary resistance as well as those events which are linked to the MLN4924 905579-51-3 eventual development of acquired resistance. Consistent with other types of cancer, primary resistance to temsirolimus is found in a subset of these cell lines. Our data suggest that primarily resistant cells lack robust Akt signaling, are unable to phosphorylate Akt at baseline, and express PTEN. In contrast, the most sensitive cell lines have lost PTEN expression and have high baseline phosphorylation of Akt. Our data demonstrate that in these cells, temsirolimus treatment promotes a further increase in Akt phosphorylation, indicating that signaling through the prosurvival PI3K/Akt pathway is likely how these endometrial cancer cell lines eventually circumvent mTOR inhibition. These results are consistent with previous reports in other types of cancers documenting compensatory Akt phosphorylation in response to other rapalogs. This has been observed in xenograft models of lung canceras well as in advanced colon and breast cancer tissues following rapalog therapy. The elevated Akt phosphorylation is thought to be a predominant driving force in resistance to temsirolimus treatment in these cancers. To overcome  resistance, we adopted a combination strategy. Dual treatment with temsirolimus and the PI3K inhibitor ZSTK474 or the PI3K/mTOR inhibitor BEZ235 overcame the temsirolimus-induced Akt hyper-phosphorylation, which is a marker for developing acquired resistance.
resistance, we adopted a combination strategy. Dual treatment with temsirolimus and the PI3K inhibitor ZSTK474 or the PI3K/mTOR inhibitor BEZ235 overcame the temsirolimus-induced Akt hyper-phosphorylation, which is a marker for developing acquired resistance.
Although DMOG can also inhibit KDMs this inhibition does not appear to be critical for the observed radioprotection
Histone methylation plays a key role in regulating chromatin structure, transcriptional activity and the DNA damage response. The importance of histone methylation in the DNA damage response is highlighted by the observation that inactivation of H3K9me3 methyltransferases leads to genomic instability and an inability to correctly repair DSBs caused by IR. Several KDMs, including KDM4B and KDM3A, contain Hypoxia Response Elementsand are transcriptionally activated by Hif1a under hypoxic conditions, suggesting that histone methylation may decrease under hypoxic conditions. Accordingly, we examined how the stabilization of Hif1a by DMOG impacts methylation of H3K9me3. In figure 4A, exposure of cells to DMOG rapidly increased H3K9me3 in cells. This was somewhat unexpected, since Hif1a transcriptionally upregulates 2 H3K9me3 specific KDMs, KDM4B and KDM3A, and should therefore decrease H3K9me3 levels. However, KDMs and the Hif1a prolylhydroxylases belong to the larger family of dioxygenases. Both the Hif1a prolylhydroxylases and KDMs utilize an Feand 2-oxoglutarate-dependent dioxygenase mechanism to either hydroxylate proline on Hif1aor remove methyl groups from methylated lysines on histone tails. DMOG, which is a 2-oxoglutarate analog, is a competitive inhibitor of the Hif1aPH and is therefore likely to inhibit KDMs as well. In figure 4B, the H3K9me3 specific demethylase KDM4A was transiently expressed in cells. Cells expressing vector showed increased H3K9me3 methylation when cells were exposed to DMOG, consistent with inhibition of endogenous KDMs. Evofosfamide 918633-87-1 Overexpression of KDM4A resulted in almost complete loss of endogenous H3K9me3 in the cells; however, addition of DMOG to cells expressing KDM4A restored H3K9me3 levels to near MK-2206 normal. Figure 4B therefore clearly demonstrates that DMOG, in addition to increasing Hif1a protein levels, can also inhibit endogenous KDMs and therefore increase levels of H3K9me3 in the cells. To further analyze how DMOG may regulate H3K9me3 levels, we also determined if Hif1a could increase expression of Suv39h1, a key H3K9me3 methyltransferase. Surprisingly, figure 4C and 4D demonstrate that both DMOG and, to a lesser extent, CoCl2, increased expression of the Suv39h1 methyltransferase. Further, the increase in Suv39h1 protein closely followed the dosedependentand time-dependentincrease in Hif1a levels caused by DMOG. Importantly, both basal and DMOG dependent induction of Suv39h1 were abolished in cells lacking Hif1a, indicating that Hif1a directly regulates the levels of Suv39h1 in cells. Figure 4 therefore demonstrates that DMOG can influence H3K9me3 levels by directly inactivating H3K9 demethylasesas well as upregulating H3K9 methyltransferases through a Hif1a dependent mechanism  Figure 4C�CE). The previous results indicate that DMOG may influence radiosensitivity through 2 distinct pathways. First, DMOG can directly inhibit KDMs, increasing H3K9me3 levels by a mechanism which is independent of Hif1a. Second, DMOG can directly inhibit Hif1a-prolylhydroxylases, leading to stabilization and accumulation of transcriptionally active Hif1a. To determine the relative contributions of these 2 pathways to the ability of DMOG to function as a radioprotector, we examined how silencing of Hif1a with shRNA impacted radioprotection. Figure 5A demonstrates that DMOG increased radioresistance in MEFs expressing a non-specific shRNA, but this effect was lost when Hif1a was silenced. The ability of DMOG to function as a radioprotector therefore requires Hif1a, indicating that DMOG exerts its primary effect on radiosensitivity through inhibition of the Hif1a-prolylhydroxylase and accumulation of Hif1a.
Figure 4C�CE). The previous results indicate that DMOG may influence radiosensitivity through 2 distinct pathways. First, DMOG can directly inhibit KDMs, increasing H3K9me3 levels by a mechanism which is independent of Hif1a. Second, DMOG can directly inhibit Hif1a-prolylhydroxylases, leading to stabilization and accumulation of transcriptionally active Hif1a. To determine the relative contributions of these 2 pathways to the ability of DMOG to function as a radioprotector, we examined how silencing of Hif1a with shRNA impacted radioprotection. Figure 5A demonstrates that DMOG increased radioresistance in MEFs expressing a non-specific shRNA, but this effect was lost when Hif1a was silenced. The ability of DMOG to function as a radioprotector therefore requires Hif1a, indicating that DMOG exerts its primary effect on radiosensitivity through inhibition of the Hif1a-prolylhydroxylase and accumulation of Hif1a.
Qualitative evaluation in our system confirmed that cells on TCPS proliferated at a higher rate than those on soft substrates
MK2 is known to regulate the expression of several proinflammatory cytokines, however we focused on the down regulation of TNF-a protein expression as a marker of YARA activity. In previous studies we have noted that cytokine expression is down regulated at 6 hours and continues to be down regulated at 24 hours with the difference between expression in control and treated cells maximized at the later time point. We believe that the prolonged ability of the peptide to suppress cytokine protein expression is due to uptake into and prolonged residence time in caveolae. This is consistent with work by Kim, et al. where they saw prolonged retention of TAT-M13-phage in caveolae, and is subject to further ICI 182780 company investigation. The concentration required to demonstrate efficacy in this cell line was equivalent to that observed in animal models when cells are seeded at low densities on polyacrylamide substrates. The importance of cell density must not be overlooked. Previous studies in our laboratory indicated that confluent mesothelial cells seeded on GW786034 tissue culture plastic exhibited a decrease in cytokine production only when treated with very high concentrations of peptide. However, when cells were seeded at a much lower density on tissue culture plastic, there was evidence of efficacy at 100 mM. Mesothelial cells were initially cultured and evaluated for efficacy at such a high density because it was thought that the cell-cell contact was best for mimicking the in vivo environment where the cells form a monolayer called the mesothelium.  However, based on the results presented herein, this assumption is clearly incorrect. Cell context is critical to cell behavior, and cells change their genetic profile base on cellcell contacts, how close to the edge of a culture they are, and even cell density. Similar to other mammalian cells, mesothelial cells have the ability to change phenotype, and it is possible that at high seeding concentrations the mesothelial cells are differentiating and are thus not as responsive to YARA. It is also possible that the rate of endocytosis is decreased at high seeding densities, due to phenotypic and metabolic changes at high cell densities. This latter possibility is consistent with the studies of Snijder, et al. where they used both modeling and experiment to show that the rate of endocytosis changed with cell density, and how close the edge of a cell islet the individual cell was. Thus, phenotypic changes due to cell density may explain why such variability is observed in YARA efficacy when cells are seeded at high concentrations, and further explain the high concentration of YARA required to observe efficacy at confluence. Both substrate stiffness and the extracellular matrix have previously been shown to regulate non-viral gene delivery. In these cases, the gene transfer efficiency of plasmidDNA increased with increasing density of RGD peptides, with fibronectin coating and with increasing stiffness. The enhanced efficiency of uptake with higher density of RGD peptides and increased stiffness is attributed to an increase in cellular proliferation, while the enhanced uptake with fibronectin coating is attributed to the promotion of endocytic uptake by clathrin-mediated endocytosis. That stiffness has an opposing role in plasmid-DNA uptake compared to our MK2-inhibitor peptide is not surprising. PlasmidDNA is often cited as being taken up through clathrin-mediated endocytosis, while the MK2-inhibitor in mesothelial cells gets taken up mainly through caveolae-mediated endocytosis. While proliferation can be important for uptake, as seen in the case of plasmid DNA-uptake, it does not appear to have the same effect in the case of YARA uptake.
However, based on the results presented herein, this assumption is clearly incorrect. Cell context is critical to cell behavior, and cells change their genetic profile base on cellcell contacts, how close to the edge of a culture they are, and even cell density. Similar to other mammalian cells, mesothelial cells have the ability to change phenotype, and it is possible that at high seeding concentrations the mesothelial cells are differentiating and are thus not as responsive to YARA. It is also possible that the rate of endocytosis is decreased at high seeding densities, due to phenotypic and metabolic changes at high cell densities. This latter possibility is consistent with the studies of Snijder, et al. where they used both modeling and experiment to show that the rate of endocytosis changed with cell density, and how close the edge of a cell islet the individual cell was. Thus, phenotypic changes due to cell density may explain why such variability is observed in YARA efficacy when cells are seeded at high concentrations, and further explain the high concentration of YARA required to observe efficacy at confluence. Both substrate stiffness and the extracellular matrix have previously been shown to regulate non-viral gene delivery. In these cases, the gene transfer efficiency of plasmidDNA increased with increasing density of RGD peptides, with fibronectin coating and with increasing stiffness. The enhanced efficiency of uptake with higher density of RGD peptides and increased stiffness is attributed to an increase in cellular proliferation, while the enhanced uptake with fibronectin coating is attributed to the promotion of endocytic uptake by clathrin-mediated endocytosis. That stiffness has an opposing role in plasmid-DNA uptake compared to our MK2-inhibitor peptide is not surprising. PlasmidDNA is often cited as being taken up through clathrin-mediated endocytosis, while the MK2-inhibitor in mesothelial cells gets taken up mainly through caveolae-mediated endocytosis. While proliferation can be important for uptake, as seen in the case of plasmid DNA-uptake, it does not appear to have the same effect in the case of YARA uptake.
The best compounds fully inhibited hABHD12 with the IC50 value in mind that the solubility of allobetulin derivative
Finally, poor inhibitory activity of the isoxazole 38 allowed us to conclude that a functional group with a hydrogen bond donor or acceptor at carbon 3 was crucial for ABHD12 inhibitory activity. In summary, the above-described SAR studies allowed us to identify four key determinants for hABHD12 inhibition potency and efficacy. These key features played an important role in building a pharmacophore model of ABHD12 that is described later in this chapter. Shape complementarity of the triterpene skeleton accompanied with four axial methyl substituents likely play an important role in inhibitor binding. Additional double bonds within the skeleton affect the overall planar shape of triterpene scaffold, leading to total lack of inhibitory activity. Carboxyl group at position 17 in triterpene core structure is of crucial importance, as basically any modification at this position reduced, or fully eliminated inhibitory activity. It is known from previous studies that the carboxyl group at this position is crucial also in many other biological Nutlin-3 structure targets. Small, hydrophobic substituents at the position 4 are required, 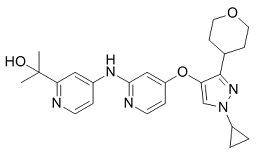 as asiatic acid and hederagenin did not inhibit hABHD12. As summarized in Figure S3, hydrogen bond donor or acceptor attached to position 3 was a key feature required for high inhibitory potency. This was further illustrated by pyridine and pyrazine derivatives 40�C42. GDC-0941 compounds 41 and 42 have a nitrogen at this position and show moderate inhibitory activity. In contrast, no atom capable of hydrogen bonding is present in the compound 40, causing dramatic decrease of inhibitory activity. A good pair of compounds for comparison was 37 and 38. The pyrazole derivative 37 was one of the most potent compounds in the series. On the other hand, when an aromatic nitrogen was replaced by an oxygen, a complete loss of the inhibitory activity was observed. This was due to the fact that oxygen within an aromatic ring cannot form hydrogen bond. The most promising synthetic compounds in the series were compounds 33 and 34 which both have an indole ring attached to ring A, and thus, a nitrogen atom at this crucial hydrogen bonding position. The compound 34 has an electron donating methoxy group at the indole ring which may cause tighter interaction between indole nitrogen and amino acid residues of the enzyme. In addition, good inhibitory activity of this methoxy derivative also implies that there is additional space for bulkier substituents in this direction. Strength of a hydrogen bond may also explain why these compounds were equally potent in inhibiting hABHD12 activity but their maximal inhibition was significantly different. Similar trend in efficacy was observed with the compounds 41 and 42, i.e. additional nitrogen in pyrazine decreased efficacy. Collectively, the above data demonstrate the importance of a hydrogen bond donor at position 3. However, as betulinic acid that can act both as a hydrogen bond acceptor and a donor, and compound 18 that is a hydrogen bond acceptor, both showed good inhibitory activity, we were able to conclude that both hydrogen bond donor and acceptor are tolerated at this position. The key finding that both hydrogen bond donor and acceptor at position 3 are able to form good interactions with the enzyme led us to hypothesize that these interactions might involve a serine residue, possibly the catalytic serine of ABHD12, previously identified by site-directed mutagenesis. However, due to the reversible nature of triterpenoid inhibition, this hypothesis could not be experimentally tested. In conclusion, we report the discovery of the first phytocompounds and their synthetic analogues that inhibit human and mouse ABHD12. The studied compounds belong to the class of triterpenoids that are known to possess wide-ranging therapeutic effects.
as asiatic acid and hederagenin did not inhibit hABHD12. As summarized in Figure S3, hydrogen bond donor or acceptor attached to position 3 was a key feature required for high inhibitory potency. This was further illustrated by pyridine and pyrazine derivatives 40�C42. GDC-0941 compounds 41 and 42 have a nitrogen at this position and show moderate inhibitory activity. In contrast, no atom capable of hydrogen bonding is present in the compound 40, causing dramatic decrease of inhibitory activity. A good pair of compounds for comparison was 37 and 38. The pyrazole derivative 37 was one of the most potent compounds in the series. On the other hand, when an aromatic nitrogen was replaced by an oxygen, a complete loss of the inhibitory activity was observed. This was due to the fact that oxygen within an aromatic ring cannot form hydrogen bond. The most promising synthetic compounds in the series were compounds 33 and 34 which both have an indole ring attached to ring A, and thus, a nitrogen atom at this crucial hydrogen bonding position. The compound 34 has an electron donating methoxy group at the indole ring which may cause tighter interaction between indole nitrogen and amino acid residues of the enzyme. In addition, good inhibitory activity of this methoxy derivative also implies that there is additional space for bulkier substituents in this direction. Strength of a hydrogen bond may also explain why these compounds were equally potent in inhibiting hABHD12 activity but their maximal inhibition was significantly different. Similar trend in efficacy was observed with the compounds 41 and 42, i.e. additional nitrogen in pyrazine decreased efficacy. Collectively, the above data demonstrate the importance of a hydrogen bond donor at position 3. However, as betulinic acid that can act both as a hydrogen bond acceptor and a donor, and compound 18 that is a hydrogen bond acceptor, both showed good inhibitory activity, we were able to conclude that both hydrogen bond donor and acceptor are tolerated at this position. The key finding that both hydrogen bond donor and acceptor at position 3 are able to form good interactions with the enzyme led us to hypothesize that these interactions might involve a serine residue, possibly the catalytic serine of ABHD12, previously identified by site-directed mutagenesis. However, due to the reversible nature of triterpenoid inhibition, this hypothesis could not be experimentally tested. In conclusion, we report the discovery of the first phytocompounds and their synthetic analogues that inhibit human and mouse ABHD12. The studied compounds belong to the class of triterpenoids that are known to possess wide-ranging therapeutic effects.