The tumor cell specific killing observed by the depletion of DDK has aroused interest as a pharmaceutical target for cancer therapy. Efforts by multiple pharmaceutical companies have led to a number of small molecule DDK inhibitors. The first well-characterized DDK inhibitor was a pyrrolopyridinone molecule. It is a potent DDK inhibitor with an IC50 of 10 nM using purified kinase. PHA767491 is also an effective cell growth inhibitor, with an average IC50 =3.14 mM among 61 tumor cell lines. PHA-767491 also inhibits purified Cdk9 with an IC50 of 34 nM but is a much less potent inhibitor of many other kinases tested. Hence PHA-767491 is a dual DDK/Cdk9 inhibitor. Recent studies have suggested that inhibition of Cdk9, a kinase that targets RNA Polymerase II, might enhance the apoptotic response induced by PHA-767491 in some cell lines. Modifications of this compound led to the identification of several other potent inhibitors of DDK with some exhibiting superior selectivity and sensitivity. XL413, a structurally distinct DDK inhibitor, is a benzofuropyrimidinone based compound with a reported IC50 of 3.4 nM against purified DDK 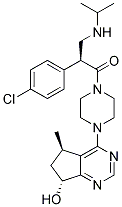 and inhibits cell-proliferation of Colo-205 cells with an IC50 of 2.69 mM. It was also highly selective for DDK when GSK2118436 tested against a panel of 100 kinases. The increased activity and selectivity of XL413 over PHA767491 was rationalized by the crystal structure of DDK in complex with the two DDK inhibitors. One reason XL413 might be a more specific inhibitor is that it made contacts with three of the most variant residues in the kinase active site when compared to PHA-767491, which interacted with two of these residues. It was therefore unexpected to find that XL413 was not a particularly potent cell growth inhibitor in most of the cell lines we tested, since Cdc7 is essential for cell cycle progression. XL413 Temozolomide customer reviews inhibited proliferation and induced apoptosis in Colo-205 cells as shown previously but had limited activity in 9 other tumor cell lines tested. Although both compounds are comparable biochemical DDK inhibitors, PHA-767491 exhibited superior activity to XL413 in cell lines. Analysis of DDK-specific Mcm2 phosphorylation levels suggests that XL413 might have poor bioavailability in these and other cancer cell lines. To aid in the development of additional DDK inhibitors, we tested whether known protein kinase inhibitors exhibited cross-reaction with DDK. We screened,400 compounds using a thermal stability shift assay and identified 12 molecules that shifted the thermal stability of DDK, several with divergent chemical scaffolds and with nearly equivalent potency as PHA-767491. These compounds are therefore unlikely to be highly specific for a single target. Our data highlight the opportunity to design additional specific, biologically active DDK inhibitors for use as chemotherapeutic agents. Small molecule inhibitors have been successfully employed both in the clinic and laboratory. Despite being initially regarded as too non-specific for deployment in therapy, small molecule kinase inhibitors have emerged as frontrunners in drug development, especially against cancer. Clinically useful molecules are often called ��drugs’ while the ones used for studying protein functions in the laboratory are called ��chemical probes’. Both the groups share a basic requirement of high potency against the target of interest. While drugs need to act effectively against the targeted disease and exhibit good pharmacokinetic properties in a physiological setting, for chemical probes target specificity is of paramount importance. Small molecule inhibitors of DDK are attractive both as drugs as well as chemical probes. Since the initial description of the tumor specific cell killing observed in response to depletion of DDK, several DDK inhibitors have been synthesized. Very different families of chemical moieties have been shown to exhibit DDK inhibitory activities. Nerviano Medical Sciences, Roche, Abbot, Exelixis, and Amgen have developed and characterized DDK inhibitors.
and inhibits cell-proliferation of Colo-205 cells with an IC50 of 2.69 mM. It was also highly selective for DDK when GSK2118436 tested against a panel of 100 kinases. The increased activity and selectivity of XL413 over PHA767491 was rationalized by the crystal structure of DDK in complex with the two DDK inhibitors. One reason XL413 might be a more specific inhibitor is that it made contacts with three of the most variant residues in the kinase active site when compared to PHA-767491, which interacted with two of these residues. It was therefore unexpected to find that XL413 was not a particularly potent cell growth inhibitor in most of the cell lines we tested, since Cdc7 is essential for cell cycle progression. XL413 Temozolomide customer reviews inhibited proliferation and induced apoptosis in Colo-205 cells as shown previously but had limited activity in 9 other tumor cell lines tested. Although both compounds are comparable biochemical DDK inhibitors, PHA-767491 exhibited superior activity to XL413 in cell lines. Analysis of DDK-specific Mcm2 phosphorylation levels suggests that XL413 might have poor bioavailability in these and other cancer cell lines. To aid in the development of additional DDK inhibitors, we tested whether known protein kinase inhibitors exhibited cross-reaction with DDK. We screened,400 compounds using a thermal stability shift assay and identified 12 molecules that shifted the thermal stability of DDK, several with divergent chemical scaffolds and with nearly equivalent potency as PHA-767491. These compounds are therefore unlikely to be highly specific for a single target. Our data highlight the opportunity to design additional specific, biologically active DDK inhibitors for use as chemotherapeutic agents. Small molecule inhibitors have been successfully employed both in the clinic and laboratory. Despite being initially regarded as too non-specific for deployment in therapy, small molecule kinase inhibitors have emerged as frontrunners in drug development, especially against cancer. Clinically useful molecules are often called ��drugs’ while the ones used for studying protein functions in the laboratory are called ��chemical probes’. Both the groups share a basic requirement of high potency against the target of interest. While drugs need to act effectively against the targeted disease and exhibit good pharmacokinetic properties in a physiological setting, for chemical probes target specificity is of paramount importance. Small molecule inhibitors of DDK are attractive both as drugs as well as chemical probes. Since the initial description of the tumor specific cell killing observed in response to depletion of DDK, several DDK inhibitors have been synthesized. Very different families of chemical moieties have been shown to exhibit DDK inhibitory activities. Nerviano Medical Sciences, Roche, Abbot, Exelixis, and Amgen have developed and characterized DDK inhibitors.