The preclinical model used in this study reproduces thus clinical situations in large chondrosarcoma. This suggests that everolimus could be worth exploring as adjuvant treatment at least in patients with grade 2 or higher chondrosarcoma. Whether everolimus would be able to show the same antitumor activity in all chondrosarcoma subtypes will be tested in a prospective randomized trial scheduled to be activated in 2012 in the French Sarcoma Group. Although everolimus as monotherapy showed a strong antitumor effect and did not induce an increase in phosphorilated Akt in our chondrosarcoma model one cannot put aside the possibility that resistance could emerge in response to long term mTORC1 inhibition.ItisknownthatblockadeofmTORsignalingbyrapalogs leads to loss of ABT-199 feedback inhibition on Akt. That could potentially result in increased cell survival and resistance to cancer therapy. To prevent such resistance mechanism and additionally improve everolimus therapeutic efficiency everolimus-based combination therapy could 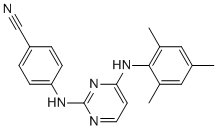 be envisionned. Such dual targeted approaches targeting mTOR and Akt, or mTOR and PI3K have proven to be pertinent in preclinical models and one has reached the clinical phase in patients with advanced sarcomas and other solid tumors. Another possible combination could be to add a bone remodelling agent to everolimus. Indeed, the combination of zoledronate to everolimus was effective in inhibiting tumor progression and in protecting bone in murine osteosarcoma model. The latter effect being the result of zoledronate rather than the one of everolimus. Like osteosarcoma, chondrosarcoma is characterized by a tumor-induced osteolysis; moreover, zoledronate has already proven to be an efficient agent in the same chondrosarcoma model. Thus it seems pertinent to hypothesize that the combination of everolimus to zoledronate could be efficient in this tumor. Such combined therapies are worth exploring in preclinical settings. In conclusion, the present results show that everolimus would be an effective antitumor agent in chondrosarcoma. Besides, the inhibition of tumor regrowth following surgery suggests that everolimus could be used as adjuvant long-term therapy in chondrosarcoma patients following surgery. These results open the way to new therapeutic approaches and led to a prospective phase II clinical trial initiatied in the French Sarcoma Group. After infection of the host cell and liberation of the RNA genome from the protecting virus particle, the viral RNA is translated into a multi-domain polyprotein that is proteolytically cleaved into ten products. The structural proteins are then used to assemble new virus particles, while the non-structural proteins participate in the replication of the viral genome. In the course of RNA replication, the viral genome is used as a template for the synthesis of negative-strand RNA, which next acts as a template for the production of positive-strand RNA. Replication is catalyzed by the NS3 helicase and the NS5B RNA-dependent RNA polymerase. The helicase represents the C-terminal portion of the NS3 protein. The NS3 helicase unwinds in an ATP-dependent manner doublestranded RNA into single strands. The shallow active site groove allows minor structural modifications to interfere with substrate binding, promoting resistance. Because NS5B, the RNA-dependent RNA polymerase, misincorporates bases at a high rate, HCV constantly mutates as it replicates. The process of constant mutation leads to heterogeneous viral populations and multiple quasispecies of HCV in Z-VAD-FMK infected patients. Mutations in the viral genome cause a rapid emergence of HCV genotypes which resist therapeutic intervention and help the virus to evade.
be envisionned. Such dual targeted approaches targeting mTOR and Akt, or mTOR and PI3K have proven to be pertinent in preclinical models and one has reached the clinical phase in patients with advanced sarcomas and other solid tumors. Another possible combination could be to add a bone remodelling agent to everolimus. Indeed, the combination of zoledronate to everolimus was effective in inhibiting tumor progression and in protecting bone in murine osteosarcoma model. The latter effect being the result of zoledronate rather than the one of everolimus. Like osteosarcoma, chondrosarcoma is characterized by a tumor-induced osteolysis; moreover, zoledronate has already proven to be an efficient agent in the same chondrosarcoma model. Thus it seems pertinent to hypothesize that the combination of everolimus to zoledronate could be efficient in this tumor. Such combined therapies are worth exploring in preclinical settings. In conclusion, the present results show that everolimus would be an effective antitumor agent in chondrosarcoma. Besides, the inhibition of tumor regrowth following surgery suggests that everolimus could be used as adjuvant long-term therapy in chondrosarcoma patients following surgery. These results open the way to new therapeutic approaches and led to a prospective phase II clinical trial initiatied in the French Sarcoma Group. After infection of the host cell and liberation of the RNA genome from the protecting virus particle, the viral RNA is translated into a multi-domain polyprotein that is proteolytically cleaved into ten products. The structural proteins are then used to assemble new virus particles, while the non-structural proteins participate in the replication of the viral genome. In the course of RNA replication, the viral genome is used as a template for the synthesis of negative-strand RNA, which next acts as a template for the production of positive-strand RNA. Replication is catalyzed by the NS3 helicase and the NS5B RNA-dependent RNA polymerase. The helicase represents the C-terminal portion of the NS3 protein. The NS3 helicase unwinds in an ATP-dependent manner doublestranded RNA into single strands. The shallow active site groove allows minor structural modifications to interfere with substrate binding, promoting resistance. Because NS5B, the RNA-dependent RNA polymerase, misincorporates bases at a high rate, HCV constantly mutates as it replicates. The process of constant mutation leads to heterogeneous viral populations and multiple quasispecies of HCV in Z-VAD-FMK infected patients. Mutations in the viral genome cause a rapid emergence of HCV genotypes which resist therapeutic intervention and help the virus to evade.
Month: August 2019
For its cleavage activity in vitro NS3 requires either the fulllength NS4A extensive evaluation
As patients begin treatment, the selective pressures of anti-virals will favor drug resistant quasispecies. Mutations that confer the most severe resistance in the clinic occur where inhibitors protrude from the consensus volume defining the substrate envelope, as these changes selectively weaken inhibitor binding without compromising the substrate binding. Both FDA-approved boceprevir and telaprevir exhibit a ketoamide moiety with the catalytic serine nucleophile and these inhibitors generate a covalent, albeit reversible, enzyme-inhibitor complex. GW786034 Additional NS3/ 4A-targeting compounds, non-covalent reversible peptidomimetic macrocycle inhibitors. These macrocyclic inhibitors exhibit an overlapping, albeit distinct, resistance profile compared with FDA-approved boceprevir and telaprevir ketoamides. Because of its functional Y-27632 moa 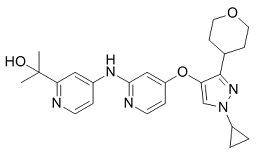 importance in the HCV life cycle, NS3/4A is an attractive anti-viral drug target. The current inhibitors can be roughly divided into two classes, macrocyclic and linear, peptidomimetic a-ketoamide derivatives. Peptidomimetic macrocyclic ciluprevir that non-covalently binds the NS3/4A active site failed clinical trials because of its cardiotoxicity. In turn, the linear peptidomimetic a-ketoamides, telaprevir and boceprevir, that bind covalently, albeit reversibly, to the active site Ser-139, have recently been approved by the FDA for clinical use. To compensate for the shallow active site groove architecture both a-ketoamides exploit interactions with catalytically non-essential amino acid residues. This results in exhibiting of a low genetic barrier for viral resistance. The association of telaprevir with NS3/ 4A involves a non-covalent binding step. This step is followed by a fast covalent bond formation resulting to the improved KI value of <40 nM. Multiple non-essential residue mutations, including, but not limited to A156F/T/V, R155K/T/Q and V36A, may rapidly lead to the telaprevir-resistant HCV, a phenomenon that has already been reported using replicon studies and murine models and, most importantly, has already been observed clinically at frequencies of 5 to 20% of the total virus population and as early as the second day after treatment initiation. To this end, we have previously demonstrated that the functional activity of the structurally similar NS2B-NS3 two-component proteinase of West Nile virus is efficiently repressed by small molecule allosteric inhibitors. Here, we employ a similar strategy to design and then test the inhibitory potency of the inhibitors that target three distinct exosites in the NS3/4A molecule. As a result, we identified novel, previously uncharacterized inhibitory scaffolds that specifically target HCV NS3/4A and the efficacy of which is not significantly affected by several common resistance mutations. HCV is a causative agent of chronic liver disease worldwide with millions of infected patients at risk of developing significant morbidity and mortality. The HCV-encoded NS3/4A is essential for viral polyprotein processing and viral replication and has long been considered a promising drug target for pharmacological intervention in HCV-infected patients. The NS3 proteinase represents the N-end180-residue, domain of the 631-residue NS3 protein. The C-end domain of NS3 encodes the ATP-dependent RNA helicase. In the course of polyprotein processing, NS3/4A cleaves the NS3-NS4A, NS4ANS4B, NS4B-NS5A and NS5A-NS5B junctions and, as a result, generates the essential late viral non-structural proteins.
importance in the HCV life cycle, NS3/4A is an attractive anti-viral drug target. The current inhibitors can be roughly divided into two classes, macrocyclic and linear, peptidomimetic a-ketoamide derivatives. Peptidomimetic macrocyclic ciluprevir that non-covalently binds the NS3/4A active site failed clinical trials because of its cardiotoxicity. In turn, the linear peptidomimetic a-ketoamides, telaprevir and boceprevir, that bind covalently, albeit reversibly, to the active site Ser-139, have recently been approved by the FDA for clinical use. To compensate for the shallow active site groove architecture both a-ketoamides exploit interactions with catalytically non-essential amino acid residues. This results in exhibiting of a low genetic barrier for viral resistance. The association of telaprevir with NS3/ 4A involves a non-covalent binding step. This step is followed by a fast covalent bond formation resulting to the improved KI value of <40 nM. Multiple non-essential residue mutations, including, but not limited to A156F/T/V, R155K/T/Q and V36A, may rapidly lead to the telaprevir-resistant HCV, a phenomenon that has already been reported using replicon studies and murine models and, most importantly, has already been observed clinically at frequencies of 5 to 20% of the total virus population and as early as the second day after treatment initiation. To this end, we have previously demonstrated that the functional activity of the structurally similar NS2B-NS3 two-component proteinase of West Nile virus is efficiently repressed by small molecule allosteric inhibitors. Here, we employ a similar strategy to design and then test the inhibitory potency of the inhibitors that target three distinct exosites in the NS3/4A molecule. As a result, we identified novel, previously uncharacterized inhibitory scaffolds that specifically target HCV NS3/4A and the efficacy of which is not significantly affected by several common resistance mutations. HCV is a causative agent of chronic liver disease worldwide with millions of infected patients at risk of developing significant morbidity and mortality. The HCV-encoded NS3/4A is essential for viral polyprotein processing and viral replication and has long been considered a promising drug target for pharmacological intervention in HCV-infected patients. The NS3 proteinase represents the N-end180-residue, domain of the 631-residue NS3 protein. The C-end domain of NS3 encodes the ATP-dependent RNA helicase. In the course of polyprotein processing, NS3/4A cleaves the NS3-NS4A, NS4ANS4B, NS4B-NS5A and NS5A-NS5B junctions and, as a result, generates the essential late viral non-structural proteins.
The molecule is associated with the intestinal alkaline phosphatas concentration is an important factor for the development of atherosclerosis
The DKO mouse, the ZDF rat, and the fructose rat model exhibited a significant increase of the plasma TG concentration and in these animals, the compounds were able to reduce plasma TG, indicating that this reduction was not model dependent. These observations do not agree with previously published observations showing that CD36 deletion in mice impairs lipoprotein lipase-mediated TG clearance and results in increased levels 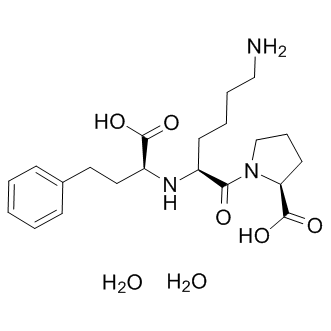 of plasma triglycerides. The present study demonstrates that an anti-CD36-ox-LDL and Fatty Acid binding activity has the capacity to reduce plasma triglycerides in rodent species. This reduction was in good agreement with the observed reduction of lipoprotein deposition in the aortic tree and the plaque growth. CD36 is implicated in lipid metabolism but has not yet been implicated in lipogenesis. Therefore it is unlikely that an inhibitor of CD36-binding may directly influence TG synthesis per se. While a pleiotropic activity of these new chemicals cannot be entirely excluded at the present time, the reason for this discrepancy could be multiple. In the present study, the mouse model was on a diet program for 12 week whereas in Febbraio’s studies, the CD36 null mice were fasted for 24 hour. Other differences may include gender and strain origin and differences in lipid metabolism. For instance, in the double CD36-ApoE knockout mice, plasma TG were significantly different in male and female mice, depending on the diet. In the present study we show that TG reduction was not affected by gender and genetic deletion. Alternatively, differences between a total disruption of the gene and a partial inhibition of the CD36 BAY-60-7550 function with an IP administration of an inhibitor can be expected. For instance, CD36 Tubacin expression in mice liver is low but the partial inhibitory activity of an administrated antagonist may be sufficient to reduce hepatic TG secretion. The published observation that heterozygotes with partial CD36 deficiency have reduced plasma TG is in agreement with our findings and supports this possibility. Increased plasma levels of TG and atherosclerosis are generally associated with impaired insulin action and glucose tolerance. Epidemiologic studies have implicated insulin resistance in both diabetes and coronary atherosclerosis. Diabetic patients have areas of lipid rich plaques with greater macrophage infiltration and many of the processes that are implicated in plaque progression are amplified by the diabetic parameters. But, the molecular links between diabetes and atherosclerosis are still missing. Glycaemia alone stimulates macrophage accumulation but fails to promote macrophage proliferation at sites of lesions. Defective insulin signaling is associated with the expression of CD36 and is enhanced via a CD36-dependent pathway. Increased CD36 expression has been shown to contribute to dyslipidemia and to be associated with insulin resistance and decreased glucose tolerance, suggesting that CD36 is involved in the physiopathology of insulin sensitivity. The present study supports this concept and shows that administration of small inhibitors of the CD36 functions improves insulin sensitivity and glucose tolerance in rodent animals. This activity was not animal model dependent and was not affected by genetic modifications. Therefore, anti-CD36 therapy may be beneficial to both atherogenic dyslipidemia and diabetes type2. CD36 is expressed in both human and rat enterocytes and has been shown to be involved in the control of intestinal cholesterol and fatty acid uptake and secretion. CD36 is expressed in the small intestine and plays an important role in chylomicron metabolism and the production of large postprandial triglyceride rich particles.
of plasma triglycerides. The present study demonstrates that an anti-CD36-ox-LDL and Fatty Acid binding activity has the capacity to reduce plasma triglycerides in rodent species. This reduction was in good agreement with the observed reduction of lipoprotein deposition in the aortic tree and the plaque growth. CD36 is implicated in lipid metabolism but has not yet been implicated in lipogenesis. Therefore it is unlikely that an inhibitor of CD36-binding may directly influence TG synthesis per se. While a pleiotropic activity of these new chemicals cannot be entirely excluded at the present time, the reason for this discrepancy could be multiple. In the present study, the mouse model was on a diet program for 12 week whereas in Febbraio’s studies, the CD36 null mice were fasted for 24 hour. Other differences may include gender and strain origin and differences in lipid metabolism. For instance, in the double CD36-ApoE knockout mice, plasma TG were significantly different in male and female mice, depending on the diet. In the present study we show that TG reduction was not affected by gender and genetic deletion. Alternatively, differences between a total disruption of the gene and a partial inhibition of the CD36 BAY-60-7550 function with an IP administration of an inhibitor can be expected. For instance, CD36 Tubacin expression in mice liver is low but the partial inhibitory activity of an administrated antagonist may be sufficient to reduce hepatic TG secretion. The published observation that heterozygotes with partial CD36 deficiency have reduced plasma TG is in agreement with our findings and supports this possibility. Increased plasma levels of TG and atherosclerosis are generally associated with impaired insulin action and glucose tolerance. Epidemiologic studies have implicated insulin resistance in both diabetes and coronary atherosclerosis. Diabetic patients have areas of lipid rich plaques with greater macrophage infiltration and many of the processes that are implicated in plaque progression are amplified by the diabetic parameters. But, the molecular links between diabetes and atherosclerosis are still missing. Glycaemia alone stimulates macrophage accumulation but fails to promote macrophage proliferation at sites of lesions. Defective insulin signaling is associated with the expression of CD36 and is enhanced via a CD36-dependent pathway. Increased CD36 expression has been shown to contribute to dyslipidemia and to be associated with insulin resistance and decreased glucose tolerance, suggesting that CD36 is involved in the physiopathology of insulin sensitivity. The present study supports this concept and shows that administration of small inhibitors of the CD36 functions improves insulin sensitivity and glucose tolerance in rodent animals. This activity was not animal model dependent and was not affected by genetic modifications. Therefore, anti-CD36 therapy may be beneficial to both atherogenic dyslipidemia and diabetes type2. CD36 is expressed in both human and rat enterocytes and has been shown to be involved in the control of intestinal cholesterol and fatty acid uptake and secretion. CD36 is expressed in the small intestine and plays an important role in chylomicron metabolism and the production of large postprandial triglyceride rich particles.
The Toll-receptor complex show molecular and functional associations with CD36 at the surface of cells
Therefore, genetic expression and molecular functions of CD36 are complex and controlled by membrane and tissue specific molecular associations and different cellular specific signaling pathways. This pleiotropic effect may reasonably well question the clinical relevance and safety of CD36. While the cellular functions of CD36 are recognized, its importance in the physiopathology is less well understood and often controversial. The role of CD36 in the formation of foam cells and the growth of atherosclerotic plaques is well documented. Yet the role of CD36 as a target to combat atherosclerosis was criticized. Similarly, evidences supporting a role of CD36 in intestinal fat absorption are accumulated, but contradictory observations have also been reported concerning its direct implication in intestinal lipid trafficking and the control of postprandial hypertriglyceridemia. For instance, CD36 is expressed all through the intestinal tract and is important for the metabolism and the secretion of chylomicron into the lymph. The molecule is required for efficient intestinal absorption of LCFA and VLCFA. Yet, CD36 deficient mice exhibit a normal level of FA absorption and gene deletion does not affect LCFA uptake and TG re-esterification in mouse jejunum. Therefore the potential of CD36 as a therapeutic target is debated. In the present paper we have identified small chemical molecules which have the capacity to inhibit the FA and ox-LDL receptor function of CD36. These inhibitors were able to rescue well characterized animal models from 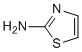 postprandial hypertriglyceridemia and atherosclerosis with a concomitant improvement of insulin resistance and Ibrutinib glucose tolerance. The CD36-inhibitor activity of this new chemical series was established on the following criteria. First, the molecules were selected for their capacity to inhibit ox-LDL binding, uptake and accumulation in THP1 cells. Furthermore, using CD36-transfected HEK cells the specificity of this inhibition for CD36 was demonstrated. Active members of this series were able to completely inhibit GDC-0941 binding and uptake to levels that were similar to the non-specific binding and uptake observed with wt cells. Second, consistent with the dual function of CD36 as a receptor for two different ligands, and the non-competitive agonist activity of these inhibitors, a similar activity on LCFA binding and uptake on both THP1 and HEK-CD36 cells was measured. These results support a receptor rather than a ligand-driven inhibition. Third, analogs of the same series with close chemical structure had no effect on these cellular functions, suggesting the existence of a structure-function relationship within the members of the series. Finally, cross-linking affinity was used to demonstrate the effect of the compounds on the molecular interaction between ox-LDL and CD36. In aggregate, these new molecules were able to inhibit the CD36 receptor function both at the cellular and the molecular levels. The first CD36 in vivo activity to be examined was its implication in the development of atherosclerosis using a well characterized animal model. A DKO mouse combining LDL-R and leptin deficiencies was used. This model exhibits high blood pressure together with increased plasma TG concentration, insulin and glucose. It develops atherosclerosis and represents a good model to study the physiopathology of the metabolic syndrome. The CD36-antagonists used in the present study were able to reduce the growth of atherosclerotic plaques at plasma concentrations compatible with the cellular activity of these molecules. This is in agreement with the fact that CD36 depleted mice are protected against atherosclerosis. Unexpectedly, a significant reduction of the plasma TG was also observed.
postprandial hypertriglyceridemia and atherosclerosis with a concomitant improvement of insulin resistance and Ibrutinib glucose tolerance. The CD36-inhibitor activity of this new chemical series was established on the following criteria. First, the molecules were selected for their capacity to inhibit ox-LDL binding, uptake and accumulation in THP1 cells. Furthermore, using CD36-transfected HEK cells the specificity of this inhibition for CD36 was demonstrated. Active members of this series were able to completely inhibit GDC-0941 binding and uptake to levels that were similar to the non-specific binding and uptake observed with wt cells. Second, consistent with the dual function of CD36 as a receptor for two different ligands, and the non-competitive agonist activity of these inhibitors, a similar activity on LCFA binding and uptake on both THP1 and HEK-CD36 cells was measured. These results support a receptor rather than a ligand-driven inhibition. Third, analogs of the same series with close chemical structure had no effect on these cellular functions, suggesting the existence of a structure-function relationship within the members of the series. Finally, cross-linking affinity was used to demonstrate the effect of the compounds on the molecular interaction between ox-LDL and CD36. In aggregate, these new molecules were able to inhibit the CD36 receptor function both at the cellular and the molecular levels. The first CD36 in vivo activity to be examined was its implication in the development of atherosclerosis using a well characterized animal model. A DKO mouse combining LDL-R and leptin deficiencies was used. This model exhibits high blood pressure together with increased plasma TG concentration, insulin and glucose. It develops atherosclerosis and represents a good model to study the physiopathology of the metabolic syndrome. The CD36-antagonists used in the present study were able to reduce the growth of atherosclerotic plaques at plasma concentrations compatible with the cellular activity of these molecules. This is in agreement with the fact that CD36 depleted mice are protected against atherosclerosis. Unexpectedly, a significant reduction of the plasma TG was also observed.
Reporters that harbour a target site for either the siRNA sense strand or passenger strand
Our observation provides additional support for the lack of incorporation of modified passenger strand. qPCR is also sometimes used to verify the inhibition of  a miRNA by transiently transfected antisense inhibitor, but this can also be problematic because the antisense inhibitor can directly inhibit the qPCR reaction. For example, in an experiment where transfection of miR-200a antisense inhibitor into MCF7 cells produced an apparent,50% decrease in miR-200a levels as measured by qPCR, we found that much of the apparent decrease in miRNA was attributable to the suppressive effect of antisense inhibitor on the PCR reaction itself. This was revealed by the addition of the same amount of antisense inhibitor directly to the cells after lysis by TRIzol, but prior to RNA extraction, which appeared to give a similar decrease in the level of miR-200a as measured by qPCR. Coupled with the fact that most of the transfected oligonucleotide is located in vesicles, this indicates that the qPCR may be largely measuring the inhibitory effect of the vesicle-associated antisense inhibitors on the qPCR, rather than its antisense activities within cells. We note that both 29-O-Methyl and LNA miRNA inhibitors are similarly subject to this phenomenon. This complements previous observations that the LNA:miRNA complex interferes with the binding of the Northern blot probe when measuring miRNA inhibition by Northern blot. Whilst miRNA mimics and antisense inhibitors are valuable tools, our observations indicate caveats to the analysis of miRNA and antisense inhibitor transfection that are apparently not universally appreciated, leading to the surprisingly frequent use in the literature of qPCR for mRNA measurement when a readout of function would be more appropriate. Better options are the use of a miRNA reporter to report the relative functional level of a miRNA, or measurement of the miRNA level following Argonaute immunoprecipitation. Progressive impairment of pancreatic Silmitasertib PKC inhibitor b-cell function and decline in b-cell mass result in relative or absolute insulin deficiency and hyperglycemia, the primary basis of all diabetic manifestations. Therefore, strategies that can induce b-cell regeneration have the potential to cure diabetes. Glucagon like peptide- 1 is released from the intestinal enteroendocrine L cells in response to nutrient ingestion. GLP-1 exerts pleiotropic actions in pancreatic islets that include stimulating glucose-dependent insulin secretion from b-cells, suppressing glucagon release from a-cells, enhancing b-cell proliferation, and preventing b-cell apoptosis. However, GLP-1 is rapidly degraded and inactivated by dipeptidylpeptidaseIV, a serine protease present in soluble form in circulation. Thus, inhibition of DPP-IV leads to an increase in circulating levels of endogenous bioactive GLP-1. DPP-IV inhibitors, such as sitagliptin, play a major role in preventing degradation of endogenous active GLP-1 and are being Dabrafenib 1195765-45-7 assessed extensively in clinical settings for their long-term efficacy in improving b-cell function in humans with type-2 diabetes mellitus. At present, DPP-IV inhibitors are the only agents in clinical use that increase endogenous GLP-1 levels. Islet b cells express several G-protein coupled receptors, one of which is the GLP-1 receptor and another one is GPR119, which is expressed predominantly in pancreatic b cells and intestinal enteroendocrine L cells. GPR119 expression has been demonstrated in isolated islets and mouse insulinoma cell lines.
a miRNA by transiently transfected antisense inhibitor, but this can also be problematic because the antisense inhibitor can directly inhibit the qPCR reaction. For example, in an experiment where transfection of miR-200a antisense inhibitor into MCF7 cells produced an apparent,50% decrease in miR-200a levels as measured by qPCR, we found that much of the apparent decrease in miRNA was attributable to the suppressive effect of antisense inhibitor on the PCR reaction itself. This was revealed by the addition of the same amount of antisense inhibitor directly to the cells after lysis by TRIzol, but prior to RNA extraction, which appeared to give a similar decrease in the level of miR-200a as measured by qPCR. Coupled with the fact that most of the transfected oligonucleotide is located in vesicles, this indicates that the qPCR may be largely measuring the inhibitory effect of the vesicle-associated antisense inhibitors on the qPCR, rather than its antisense activities within cells. We note that both 29-O-Methyl and LNA miRNA inhibitors are similarly subject to this phenomenon. This complements previous observations that the LNA:miRNA complex interferes with the binding of the Northern blot probe when measuring miRNA inhibition by Northern blot. Whilst miRNA mimics and antisense inhibitors are valuable tools, our observations indicate caveats to the analysis of miRNA and antisense inhibitor transfection that are apparently not universally appreciated, leading to the surprisingly frequent use in the literature of qPCR for mRNA measurement when a readout of function would be more appropriate. Better options are the use of a miRNA reporter to report the relative functional level of a miRNA, or measurement of the miRNA level following Argonaute immunoprecipitation. Progressive impairment of pancreatic Silmitasertib PKC inhibitor b-cell function and decline in b-cell mass result in relative or absolute insulin deficiency and hyperglycemia, the primary basis of all diabetic manifestations. Therefore, strategies that can induce b-cell regeneration have the potential to cure diabetes. Glucagon like peptide- 1 is released from the intestinal enteroendocrine L cells in response to nutrient ingestion. GLP-1 exerts pleiotropic actions in pancreatic islets that include stimulating glucose-dependent insulin secretion from b-cells, suppressing glucagon release from a-cells, enhancing b-cell proliferation, and preventing b-cell apoptosis. However, GLP-1 is rapidly degraded and inactivated by dipeptidylpeptidaseIV, a serine protease present in soluble form in circulation. Thus, inhibition of DPP-IV leads to an increase in circulating levels of endogenous bioactive GLP-1. DPP-IV inhibitors, such as sitagliptin, play a major role in preventing degradation of endogenous active GLP-1 and are being Dabrafenib 1195765-45-7 assessed extensively in clinical settings for their long-term efficacy in improving b-cell function in humans with type-2 diabetes mellitus. At present, DPP-IV inhibitors are the only agents in clinical use that increase endogenous GLP-1 levels. Islet b cells express several G-protein coupled receptors, one of which is the GLP-1 receptor and another one is GPR119, which is expressed predominantly in pancreatic b cells and intestinal enteroendocrine L cells. GPR119 expression has been demonstrated in isolated islets and mouse insulinoma cell lines.