The possibility that miRs might modulate mRNA levels and subsequent toxicity by TCDD has not been fully explored. Recently, a few studies have begun exploring such mechanisms. One study reported that miR-27b related to AhR-regulated genes increased CYP1B1 levels. In another study, it was noted that treatment with TCDD in vivo caused few changes in miR levels in mouse or rat livers, and those changes that were statistically significant were of modest magnitude. These data are consistent with our studies where we noted that the magnitude of change in miR expression following TCDD treatment in most Tulathromycin B instances was 1.5 to 2 fold and only a few miRs showed 3�C8 fold change. The fact that liver may be more refractory was also indicated in another study in which it was noted that AhR activation by benzopyrene did not cause significant changes in miRs of the liver but altered the miR profiles in the lung. The miRs that were altered by BaP were involved in immune response, cell proliferation and cell cycle. Thus, it is likely that the AhR-agonist mediated changes in miRs may be organ-specific. While, the immunotoxic effects of prenatal exposure to TCDD on fetal thymocytes have been well characterized, there are no reports on such effects of TCDD on miR profiles. Understanding the role of various miRs in neonatal mice post-TCDD exposure may shed light on the “fetal basis of adult disease” hypothesis. This hypothesis proposes that many chronic diseases including autoimmune diseases during adult stage of life may be the result of prenatal exposure to nutritional, environmental or other forms of stress. In this study, therefore, we sought to examine miR profile in fetuses post-TCDD exposure. The cluster analysis data of miRs showed that TCDD caused significant changes in miR expression profile in fetal thymi when compared to vehicle-treated thymi. Of the miRs screened, 78 miRs were altered more than 1.5 fold and 28 miRs were altered two fold or more, post-TCDD exposure. We further validated the expression profile of some select miRs by performing Real-Time PCR. All the miRs that we analyzed by Real-Time PCR corroborated the data obtained from miR array analysis. Furthermore, the relationship of miRs and their target gene expression was also verified. For example, miRs that showed highly complementary sequence with 39UTR of AhR, CYP1A1, Fas, and FasL genes were downregulated by TCDD in fetal thymi and the data obtained from RT-PCR showed upregulated expression of the above genes in fetal thymi post-TCDD exposure. such as breast, cartilage, endothelial cells, embryonic tissues, etc. These downregulated miRs have been shown to control genes that are involved in various physiological functions in these tissues. Previous studies from our laboratory have demonstrated that TCDD-induced thymic atrophy in the adult and fetus may result, at least in part, from induction of apoptosis. We have also reported that such Folinic acid calcium salt pentahydrate apoptosis may be induced through the extrinsic pathway by the induction of Fas and FasL in thymocytes. Also miR200a has been shown to regulate apoptosis, whereas miR-491 has been shown to induce apoptosis by targeting Bcl-xL gene. Thus, these miRs may directly/indirectly be involved in apoptosis of thymic 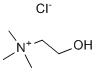 cells leading to thymic atrophy. TCDD has also been shown to cause cancer in various species and it is also considered to be a potential carcinogen in humans. There are reports demonstrating that TCDD exposure of mice triggers cutaneous papillomas and squamous cell carcinoma. In another report, prenatal TCDD exposure of rats was shown to make them susceptible to breast cancer. TCDD has also been shown to promote liver cancer. miRs have been shown to influence signaling pathways leading to development of various types of cancer.
cells leading to thymic atrophy. TCDD has also been shown to cause cancer in various species and it is also considered to be a potential carcinogen in humans. There are reports demonstrating that TCDD exposure of mice triggers cutaneous papillomas and squamous cell carcinoma. In another report, prenatal TCDD exposure of rats was shown to make them susceptible to breast cancer. TCDD has also been shown to promote liver cancer. miRs have been shown to influence signaling pathways leading to development of various types of cancer.
Category: MAPK Inhibitor Library
An inherent assumption in our method is that such TFBSs will be enriched
Since sequences in the background set used for comparison also contain RCGTG motifs, this enrichment likely arises from the well known preference for A versus G in the first position of the HIF Orbifloxacin binding consensus. These results collectively suggest that several additional transcription factors could influence HIF transcriptional activity. Importantly, we noted that most of the enriched TFBSs corresponded to stress-responsive transcription factors. Varied stress-responsive TFs have been shown to coordinately regulate the same genes, 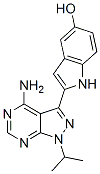 and indeed several transcription factors are activated by the same stresses in mammalian cells. However, it is unclear whether this cooperation among stressresponsive pathways translates at the genomic level. In order to evaluate the functional significance of the TFBSs enriched in core HIF binding regions, we carried out an experimental validation by disrupting selected sequences in bona fide HIF-responsive promoters. Importantly, no experimental confirmation had been attempted on previously reported predictions, and therefore the biological significance of those findings remained unclear. In spite of being limited to three selected promoters, our results clearly indicate that, compared to control mutations, alteration of binding sequences of transcription factors enriched in HIF binding regions, and different from HIFs themselves, have a specific effect on the transcriptional activation of HIF-responsive promoters. In particular, we found negative effects on hypoxic induction of LDHA and GYS1 promoters upon disruption of CREB and CEBPB binding sites proximal to the HRE, whereas mutation of an AP1 site proximal to the CA9 HRE led to a slightly augmented hypoxic induction of the promoter. In agreement with our results, mutation of the same CREB binding site was been previously shown to alter LDHA hypoxic induction. Interestingly, USF binding to a palindrome CACGTG HRE in the LDHA promoter was suggested to complement HIF binding. However, our results do not allow us to corroborate these findings, as mutation of this HRE was not evaluated in our experiments. Furthermore, hypoxic CA9 Catharanthine sulfate expression has been linked to cooperation between AP1 family member ATF4 and HIF1a. In this study, ATF4 overexpression led to an augmented CA9 induction in hypoxia, with reduced hypoxic expression of CA9 being observed upon ATF4 knock-down. Chromatin immunoprecipitation experiments mapped ATF4 binding to the 21400/21000 region of the CA9 promoter, which falls outside of the promoter region employed in our experiments. Nevertheless, the apparent paradox with our results argues for careful interpretation of the role of AP1 in the HIF transcriptional response. In fact, both positive and negative effects of AP1 have been reported on hypoxic gene expression and, given the number of AP1 family members, these probably arise from compositional differences in AP1 complexes. Importantly, the effects observed upon mutation of CREB, CEBPB or AP1 binding sites were always moderate when compared to mutation of the HIF binding consensus RCGTG, suggesting that rather than being an absolute requirement for hypoxic induction, the integrity of these neighboring TFBSs fine-tunes the HIF-mediated transcriptional response. Thus, it is possible that multiple independent factors contribute, in an additive fashion, to HIF-mediated transcription. This model could also explain why we found a relatively large number of enriched TFBSs in HIF binding regions, but all of them sharing a modest statistical significance. On the whole, these observations indicate that several of the enriched TFBSs identified in our approach are of functional relevance for HIF-mediated transcription. Nevertheless, it should be noted that other TFs for which collaboration with HIFs has been previously suggested are not recovered as enriched in our approach.
and indeed several transcription factors are activated by the same stresses in mammalian cells. However, it is unclear whether this cooperation among stressresponsive pathways translates at the genomic level. In order to evaluate the functional significance of the TFBSs enriched in core HIF binding regions, we carried out an experimental validation by disrupting selected sequences in bona fide HIF-responsive promoters. Importantly, no experimental confirmation had been attempted on previously reported predictions, and therefore the biological significance of those findings remained unclear. In spite of being limited to three selected promoters, our results clearly indicate that, compared to control mutations, alteration of binding sequences of transcription factors enriched in HIF binding regions, and different from HIFs themselves, have a specific effect on the transcriptional activation of HIF-responsive promoters. In particular, we found negative effects on hypoxic induction of LDHA and GYS1 promoters upon disruption of CREB and CEBPB binding sites proximal to the HRE, whereas mutation of an AP1 site proximal to the CA9 HRE led to a slightly augmented hypoxic induction of the promoter. In agreement with our results, mutation of the same CREB binding site was been previously shown to alter LDHA hypoxic induction. Interestingly, USF binding to a palindrome CACGTG HRE in the LDHA promoter was suggested to complement HIF binding. However, our results do not allow us to corroborate these findings, as mutation of this HRE was not evaluated in our experiments. Furthermore, hypoxic CA9 Catharanthine sulfate expression has been linked to cooperation between AP1 family member ATF4 and HIF1a. In this study, ATF4 overexpression led to an augmented CA9 induction in hypoxia, with reduced hypoxic expression of CA9 being observed upon ATF4 knock-down. Chromatin immunoprecipitation experiments mapped ATF4 binding to the 21400/21000 region of the CA9 promoter, which falls outside of the promoter region employed in our experiments. Nevertheless, the apparent paradox with our results argues for careful interpretation of the role of AP1 in the HIF transcriptional response. In fact, both positive and negative effects of AP1 have been reported on hypoxic gene expression and, given the number of AP1 family members, these probably arise from compositional differences in AP1 complexes. Importantly, the effects observed upon mutation of CREB, CEBPB or AP1 binding sites were always moderate when compared to mutation of the HIF binding consensus RCGTG, suggesting that rather than being an absolute requirement for hypoxic induction, the integrity of these neighboring TFBSs fine-tunes the HIF-mediated transcriptional response. Thus, it is possible that multiple independent factors contribute, in an additive fashion, to HIF-mediated transcription. This model could also explain why we found a relatively large number of enriched TFBSs in HIF binding regions, but all of them sharing a modest statistical significance. On the whole, these observations indicate that several of the enriched TFBSs identified in our approach are of functional relevance for HIF-mediated transcription. Nevertheless, it should be noted that other TFs for which collaboration with HIFs has been previously suggested are not recovered as enriched in our approach.
Based on recent single cargo translocation initial translocation of prophylactic efficacy of LdTPI-DNA
The Folinic acid calcium salt pentahydrate nuclear pore complex is a supramolecular protein assembly forming a highly selective channel embedded in the nuclear membrane. It regulates bidirectional nucleo-cytoplasmic transport for a large range of proteins and complexes too large to diffuse freely through the NPC. They are composed of numerous copies of,30 different nucleoporins, which have a well-assigned localisation, function and half-life, and are present as multiples of eight reflecting the highly conserved eight-fold axial symmetry of NPCs. The central substructure of the NPC is composed of transmembrane Nups that anchor the NPC to the nuclear envelope, scaffold Nups that constitute cornerstones during NPC biogenesis, and FG-Nups so-called because they contain extensive repeats of phenylalanine-glycine domains that form an unstructured mesh at the centre of the channel. Nup358/ RanBP2 and Nup214/CAN have been mapped exclusively to the cytoplasmic side of the NPC, where 50�C100 nm long flexible cytoplasmic filaments radiate from the NPC into the cytoplasm. Nup358/RanBP2 has been reported to be the major component of the cytoplasmic NPC filaments. Nup98 is a symmetrical nucleoporin, located on both the cytoplasmic and nuclear sides of the NPC. On the nuclear side of the NPC, Nups such as Nup153 and Nup98 associate with the nuclear basket and with the chromatin both in proximity of and away from the NPC. Many viruses depend on access to the nuclear compartment for replication and have evolved unique strategies to translocate into the nucleus. Retroviruses such as Murine Leukaemia Virus enter the nucleus during mitotic nuclear membrane disassembly, however other viruses such as herpesviruses and adenoviruses dock their capsids at the NPC and release their genome into the nucleus, while still others enter in the nucleus with their capsid. The Human Immunodeficiency Virus type 1, contrary to other orthoretroviruses, has evolved the ability to infect non-dividing cells through active nuclear Catharanthine sulfate import of its genome across the intact nuclear membrane through the NPC. Although several viral elements have been proposed to act as determinants of HIV-1 nuclear import, most notably integrase and the central DNA Flap, it is commonly accepted that HIV-1 depends on host cell proteins to achieve translocation. However, increasing data supports the presence of HIV-1 capsid at the nuclear pore and/or acting as a determinant of HIV-1 nuclear import. Uncoating certainly also occurs during cytoplasmic transport, and possibly accounts for the majority of incoming viral complexes. However, these may correspond to viral complexes destined for or undergoing degradation, for instance following entry by endocytosis. Our work shows that HIV-1 capsid interacts with Nup358/RanBP2 and that depletion of Nup358/RanBP2 impairs arrival of HIV-1 complexes at the nuclear envelope, thus confirming the presence of HIV-1 capsid cores at the nuclear membrane. However, our study does not show any effect for Nup358 in integration, since the strong reduction in proviral integration is simply due to a strong nuclear import defect and considering the exclusive cytoplasmic location of Nup358/RanBP2 we do not expect to find its potential viral partner in the nucleus. We cannot exclude that the absence of this nucleoporin could affect HIV-1 site integration but that may reflect 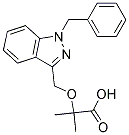 the change of chromatin environment associated with depletion of Nup358. In this work, we identified Nup358/RanBP2 as docking factor for HIV-1 capsid using independent techniques to assess docking and interaction. We are the first to identify the binding of Nup358/ RanBP2 to HIV-1 in vitro assembled CA-NC complexes suggesting that this interaction is important for HIV-1 nuclear import. Nup358/RanBP2 is one of over 20 human proteins that contain a cyclophilin-like domain.
the change of chromatin environment associated with depletion of Nup358. In this work, we identified Nup358/RanBP2 as docking factor for HIV-1 capsid using independent techniques to assess docking and interaction. We are the first to identify the binding of Nup358/ RanBP2 to HIV-1 in vitro assembled CA-NC complexes suggesting that this interaction is important for HIV-1 nuclear import. Nup358/RanBP2 is one of over 20 human proteins that contain a cyclophilin-like domain.
Clinically relevant tumor samples are therefore likely to represent the cumulative result of undirected generation of variance followed
Participate in the network transformations associated with tumorigenesis. A high level view of network rearrangements can be seen by comparing modules in AN and TU. Many significant overlaps were seen indicating that the two tissues were far from randomly organized with respect to each other. Closer examination revealed support for the disruption and creation of co-expression networks found with gene-gene differential correlation data. For example, the genes in the largest module in AN significantly overlapped the genes in eight different TU modules as well as genes that could not be placed in the TU network, consistent with disruption of the AN-turquoise module. Similarly, genes in the largest module in TU, overlapped genes in 10 AN modules as well as genes in AN-grey, consistent with ![]() the creation of TU-turquoise as part of HCC tumorigenesis. Consistent with the above interpretation of the AN and TU module overlaps was the finding that the differentially correlated genes were enriched in many of the modules. For example, 22 of the 25 AN modules and 13 of the 20 TU modules were enriched for genes containing differentially correlated genes, indicating that a majority of the subnetworks in the AN tissue representing many different biological functions were disrupted as a result of the formation and progression of the tumor, resulting in a higher level re-organization. To assess possible biological functions represented by the networks, each module was tested for over-representation of genes from individual gene ontology categories. Given the large scale reorganizations between AN and TU, GO terms that were most significantly enriched for each module compared to all other modules were defined. The purpose of this was to begin to define biological pathways that were uniquely disrupted in AN tissue and uniquely Orbifloxacin created in TU tissue. Examples of this are the findings that components of the ribosome were uniquely enriched in the TU-grey60 module, and aspects of macrophage function were uniquely enriched in the AN-coral module. In both cases these terms were not enriched in any other module of either tissue. Many unique or relative enrichments of GO terms were found for 14 of the 25 AN modules and 12 of the 21 TU modules representing a broad array of biological functions coherently represented in the networks and altered between tissues in HCC tumorigenesis. Ribosome components have been clearly implicated in tumor initiation and progression in numerous cancer types. In particular it has been argued that altered translation Cinoxacin facilitates expression of many proliferation associated genes and may also regulate the endothelial to mesenchymal transition which is thought to be important in invasion and metastasis. The enrichment of TU-survival genes in TU-grey60 and TU-lightyellow suggests that translation and ribosome biogenesis were selected for alteration during tumorigenesis, but remained rate limiting factors in progression. Copy number aberrations are widely observed in solid tumors and are likely the result of altered fidelity of DNA replication, repair, checkpoints and/or chromosome segregation. These processes leading to sCNV are by their nature intrinsic features of cancer cells and occur in an undirected manner in terms of chromosome location and direction of change. Since generation of sCNV is ongoing and will lead to neutral, increased, or decreased fitness of the corresponding cell in its environment, different derivatives will have consequently different abilities to grow and survive, therefore leading to evolution of the tumor over time.
the creation of TU-turquoise as part of HCC tumorigenesis. Consistent with the above interpretation of the AN and TU module overlaps was the finding that the differentially correlated genes were enriched in many of the modules. For example, 22 of the 25 AN modules and 13 of the 20 TU modules were enriched for genes containing differentially correlated genes, indicating that a majority of the subnetworks in the AN tissue representing many different biological functions were disrupted as a result of the formation and progression of the tumor, resulting in a higher level re-organization. To assess possible biological functions represented by the networks, each module was tested for over-representation of genes from individual gene ontology categories. Given the large scale reorganizations between AN and TU, GO terms that were most significantly enriched for each module compared to all other modules were defined. The purpose of this was to begin to define biological pathways that were uniquely disrupted in AN tissue and uniquely Orbifloxacin created in TU tissue. Examples of this are the findings that components of the ribosome were uniquely enriched in the TU-grey60 module, and aspects of macrophage function were uniquely enriched in the AN-coral module. In both cases these terms were not enriched in any other module of either tissue. Many unique or relative enrichments of GO terms were found for 14 of the 25 AN modules and 12 of the 21 TU modules representing a broad array of biological functions coherently represented in the networks and altered between tissues in HCC tumorigenesis. Ribosome components have been clearly implicated in tumor initiation and progression in numerous cancer types. In particular it has been argued that altered translation Cinoxacin facilitates expression of many proliferation associated genes and may also regulate the endothelial to mesenchymal transition which is thought to be important in invasion and metastasis. The enrichment of TU-survival genes in TU-grey60 and TU-lightyellow suggests that translation and ribosome biogenesis were selected for alteration during tumorigenesis, but remained rate limiting factors in progression. Copy number aberrations are widely observed in solid tumors and are likely the result of altered fidelity of DNA replication, repair, checkpoints and/or chromosome segregation. These processes leading to sCNV are by their nature intrinsic features of cancer cells and occur in an undirected manner in terms of chromosome location and direction of change. Since generation of sCNV is ongoing and will lead to neutral, increased, or decreased fitness of the corresponding cell in its environment, different derivatives will have consequently different abilities to grow and survive, therefore leading to evolution of the tumor over time.
The ability of the recruited cells characterized by tumor suppressor loss to contribute to high-grade glioma structures
We performed transplantation experiments using the bacTRAP system. Transplanted gliomas induced by hPDGFb-driven glioma cells in hosts with altered tumor suppressor function showed higher percentages of the overall recruitment than in the wild-type hosts, indicating that complete or partial tumor suppressor loss may enhance the ability of the cells to be  recruited. Although gliomas induced by transplantation of non-fluorescent Pten-deleted Ras-driven murine glioma cells showed recruitment of proliferating host brain cells into the glioma mass, the overall amount of recruited cells into Ras-driven tumors and the number of tumors that showed large regions of recruitment was significantly less than that seen with the hPDGFb-induced gliomas. An unbiased way to define a cell population lies in Gomisin-D identification of its gene expression signature and subsequent comparison to gene expression signatures of known normal or cancer cells to define its position on the axis of tumorigenesis. To quantify similarities and differences in the expression profiles of recruited cells and tumor cells using microarray analysis, we used the bacTRAP technology that allows immunoprecipitation of polysomes from specific cell types in vivo. RCAS/tv-a system allows to closely model the biology and histopathology of human oligodendrogliomas; although we have not performed direct comparisons between our mouse model and human glioma classifications, in many ways, hPDGFb-driven murine gliomas may mimic the proneural subclass of human GBMs. Our data suggests that olig2-expressing cells recruited in PDGF-induced murine gliomas can have similar morphologic, proliferative and functional characteristics as olig2expressing tumor cells derived from the cell-of-origin, with the caveat that a part of the similarity between the recruited and tumor olig2 cells may stem from their proliferative behavior. Extensive similarities between polysome-associated transcriptome of recruited versus glioma olig2 cells call for further evaluation of the precise definitions and criteria applied to terms “tumor” and “normal”, and indicate similar gene expression character of these cells. Furthermore, tumor suppressor loss and acquisition of mutations typically found in human gliomas allow these murine recruited cells to occupy large areas of the tumor and at times become the predominant cellular component of the glioma mass, completely independent of the cell-of-origin. Thus, the clonal expansion that overtakes glioma bulk during tumor progression need not be derived from glioma cell-of-origin. Transformation is the process of self-autonomous acquisition of the sufficient oncogenic alterations that change a normal cell into the tumor cell. Unlike epithelial cancers, the “normal” or stromal component of a glioma is composed of cells of the same lineage as the tumor cells. The inherent implication of this observation is that extracellular factors that promote glioma tumor cell growth may similarly affect glioma stroma. In our glioma model, initially normal recruited cells not derived from the glioma-initiating cellof-origin can be stimulated by hyperproduction of a growth factor receptor ligand, induced to proliferate, and driven to acquire various genetic aberrations and Cinoxacin aberrant expression profiles as gliomas progress, presumably because of the tumor microenvironment created by the progeny of the cell-of-origin. The process of acquisition and selection for such genetic and/or epigenetic alterations due to the pressures of the glioma microenvironment may be better termed “corruption”.
recruited. Although gliomas induced by transplantation of non-fluorescent Pten-deleted Ras-driven murine glioma cells showed recruitment of proliferating host brain cells into the glioma mass, the overall amount of recruited cells into Ras-driven tumors and the number of tumors that showed large regions of recruitment was significantly less than that seen with the hPDGFb-induced gliomas. An unbiased way to define a cell population lies in Gomisin-D identification of its gene expression signature and subsequent comparison to gene expression signatures of known normal or cancer cells to define its position on the axis of tumorigenesis. To quantify similarities and differences in the expression profiles of recruited cells and tumor cells using microarray analysis, we used the bacTRAP technology that allows immunoprecipitation of polysomes from specific cell types in vivo. RCAS/tv-a system allows to closely model the biology and histopathology of human oligodendrogliomas; although we have not performed direct comparisons between our mouse model and human glioma classifications, in many ways, hPDGFb-driven murine gliomas may mimic the proneural subclass of human GBMs. Our data suggests that olig2-expressing cells recruited in PDGF-induced murine gliomas can have similar morphologic, proliferative and functional characteristics as olig2expressing tumor cells derived from the cell-of-origin, with the caveat that a part of the similarity between the recruited and tumor olig2 cells may stem from their proliferative behavior. Extensive similarities between polysome-associated transcriptome of recruited versus glioma olig2 cells call for further evaluation of the precise definitions and criteria applied to terms “tumor” and “normal”, and indicate similar gene expression character of these cells. Furthermore, tumor suppressor loss and acquisition of mutations typically found in human gliomas allow these murine recruited cells to occupy large areas of the tumor and at times become the predominant cellular component of the glioma mass, completely independent of the cell-of-origin. Thus, the clonal expansion that overtakes glioma bulk during tumor progression need not be derived from glioma cell-of-origin. Transformation is the process of self-autonomous acquisition of the sufficient oncogenic alterations that change a normal cell into the tumor cell. Unlike epithelial cancers, the “normal” or stromal component of a glioma is composed of cells of the same lineage as the tumor cells. The inherent implication of this observation is that extracellular factors that promote glioma tumor cell growth may similarly affect glioma stroma. In our glioma model, initially normal recruited cells not derived from the glioma-initiating cellof-origin can be stimulated by hyperproduction of a growth factor receptor ligand, induced to proliferate, and driven to acquire various genetic aberrations and Cinoxacin aberrant expression profiles as gliomas progress, presumably because of the tumor microenvironment created by the progeny of the cell-of-origin. The process of acquisition and selection for such genetic and/or epigenetic alterations due to the pressures of the glioma microenvironment may be better termed “corruption”.