A value also detected in parental and control cells stably transfected with empty vector, that was close to that of HspB5. 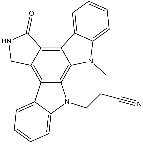 Moreover, the constitutive expression of wild type or mutant HspB5 did not modify the level of Hsp90, Hsp70, and HspB1, hence suggesting that the presence of exogenous HspB5 polypeptides inside HeLa cells was not sensed as a stress. We also tested for the presence of HspB6, a member of the family of small Hsps that can form chimeric hetero-oligomers with HspB1 in vitro. This protein was barely detectable in Neo, WT and R120G cells and was therefore not further studied. HspB1 and HspB5 appear therefore as the major interacting small Hsps that are present in Neo, WT and R120G cells. We next analyzed of the effects mediated by their interaction inside cells. Similar conclusions could be drawn when the antibody targeting HspB5 was used to perform IP. Aliquots of the total and immunodepleted pooled fractions were analyzed to verify if the immunoprecitation of the targeted protein was complete and to test the fate of the other protein partner. Since the immunodepleted supernatant fractions were devoid of the corresponding proteins, it was concluded that 100% of both HspB1 and HspB5 present in the pooled fractions were interacting and formed chimeric complexes. We then tested HspB1 and HspB5 interaction in presence of 300 mM NaCl. As seen in Fig. 2Db, interaction between HspB1 and HspB5 in the pooled fractions from WT cells was weakened by the 300 mM wash. Indeed, in the presence of high salt, a fraction of HspB5 or HspB1 corresponding partner was removed from the complex and recovered in the immunodepleted supernatant. The VE-822 in vivo phenomenon was not observed when a similar analysis was performed using the pooled fractions from R120G cells. In this case the interaction was not altered by 300 mM NaCl, hence suggesting that it was tightened by the R120G mutation. We next analyzed the resistance of Neo, WT and R120G cells to oxidative conditions since this is a common stress encountered by cells expressing HspB5, as for example when they are exposed to UV light or chronic inflammation damages. Neo, WT and R120G cells were exposed for different time periods to 60 or 100 mM of menadione, a compound that generates intracellular reactive oxygen species via redoxcycling. Subsequently, their survival was determined using crystal violet staining, clonogenic colony formation assay and phase-contrast analysis of live cells. It is seen in Fig. 3A,B, that WT cells were significantly more oxidoresistant than Neo cells while R120G cells displayed a pronounced sensitivity to menadione. Similar observations were made using Trypan blue staining of dead cells and after exposure to 100 mM of hydrogen peroxide. Morphological analysis, presented in Fig. 3C, revealed the accumulation of perinuclear vacuoles and granules in menadione-treated Neo cells that were not detected in WT cells. However, in spite of their apparent resistance to menadione, WT cells no more displayed an elongated morphology and had a more polygonal morphology; a phenomenon that could be related to the sensitivity of F-actin cytoskeleton to menadione induced oxidative stress. About half of menadione-treated R120G cells had a dying morphology: they were detached from the substratum, had a rounded appearance and were linked to each other by filamentous PF-4217903 bridges. The remaining living R120G cells were still attached and displayed a polygonal appearance. However, they had lost their dense membranous ruffles in the leading edges and were loaded with vacuoles and granules. Control experiments revealed no changes in the cellular content of HspB1 and HspB5 in response of menadione treatment as well as no stimulation of the level of two major ATP-dependent chaperones, Hsp70 and Hsp90.
Moreover, the constitutive expression of wild type or mutant HspB5 did not modify the level of Hsp90, Hsp70, and HspB1, hence suggesting that the presence of exogenous HspB5 polypeptides inside HeLa cells was not sensed as a stress. We also tested for the presence of HspB6, a member of the family of small Hsps that can form chimeric hetero-oligomers with HspB1 in vitro. This protein was barely detectable in Neo, WT and R120G cells and was therefore not further studied. HspB1 and HspB5 appear therefore as the major interacting small Hsps that are present in Neo, WT and R120G cells. We next analyzed of the effects mediated by their interaction inside cells. Similar conclusions could be drawn when the antibody targeting HspB5 was used to perform IP. Aliquots of the total and immunodepleted pooled fractions were analyzed to verify if the immunoprecitation of the targeted protein was complete and to test the fate of the other protein partner. Since the immunodepleted supernatant fractions were devoid of the corresponding proteins, it was concluded that 100% of both HspB1 and HspB5 present in the pooled fractions were interacting and formed chimeric complexes. We then tested HspB1 and HspB5 interaction in presence of 300 mM NaCl. As seen in Fig. 2Db, interaction between HspB1 and HspB5 in the pooled fractions from WT cells was weakened by the 300 mM wash. Indeed, in the presence of high salt, a fraction of HspB5 or HspB1 corresponding partner was removed from the complex and recovered in the immunodepleted supernatant. The VE-822 in vivo phenomenon was not observed when a similar analysis was performed using the pooled fractions from R120G cells. In this case the interaction was not altered by 300 mM NaCl, hence suggesting that it was tightened by the R120G mutation. We next analyzed the resistance of Neo, WT and R120G cells to oxidative conditions since this is a common stress encountered by cells expressing HspB5, as for example when they are exposed to UV light or chronic inflammation damages. Neo, WT and R120G cells were exposed for different time periods to 60 or 100 mM of menadione, a compound that generates intracellular reactive oxygen species via redoxcycling. Subsequently, their survival was determined using crystal violet staining, clonogenic colony formation assay and phase-contrast analysis of live cells. It is seen in Fig. 3A,B, that WT cells were significantly more oxidoresistant than Neo cells while R120G cells displayed a pronounced sensitivity to menadione. Similar observations were made using Trypan blue staining of dead cells and after exposure to 100 mM of hydrogen peroxide. Morphological analysis, presented in Fig. 3C, revealed the accumulation of perinuclear vacuoles and granules in menadione-treated Neo cells that were not detected in WT cells. However, in spite of their apparent resistance to menadione, WT cells no more displayed an elongated morphology and had a more polygonal morphology; a phenomenon that could be related to the sensitivity of F-actin cytoskeleton to menadione induced oxidative stress. About half of menadione-treated R120G cells had a dying morphology: they were detached from the substratum, had a rounded appearance and were linked to each other by filamentous PF-4217903 bridges. The remaining living R120G cells were still attached and displayed a polygonal appearance. However, they had lost their dense membranous ruffles in the leading edges and were loaded with vacuoles and granules. Control experiments revealed no changes in the cellular content of HspB1 and HspB5 in response of menadione treatment as well as no stimulation of the level of two major ATP-dependent chaperones, Hsp70 and Hsp90.
Category: MAPK Inhibitor Library
Changes in mRNA and protein stability alternative splicing and regulation of transcript levels
VE-822 sugarcane is a C4 monocot that stores sucrose in its stem. Commercial sugarcane varieties are the result of multiple interspecific hybridizations between Saccharum officinarum and S. spontaneum, which resulted in a highly polyploid and aneuploid genome. The high level of sucrose accumulated in sugarcane, together with its yield, make this crop an important bioenergy feedstock in a world concerned about alternatives to fossil-based fuels. Even though there is evidence that sugarcane has not reached its potential yield limit, yearly increases in sugarcane yield are low and may be plateauing, a trend that may be reversed by the introduction of new biotechnological tools. One strategy to develop new biotechnological tools to use in sugarcane improvement is to know more about its genome, gene networks and physiology. Microarrays have been a successful strategy to identify genes of interest in sugarcane. We have recently developed a new custom oligoarray with more than twenty-one thousand elements with PF-4217903 probes that hybridize to sense and antisense sugarcane EST sequences. This array identified 928 differentially expressed probes in the sense direction and 59 in the antisense direction in sugarcane subjected to water suppression, adding considerable knowledge to previous experiments that hybridized the same samples using a 1,545 elements microarray and identified 93 differentially expressed transcripts probes. Natural antisense transcripts have been shown to regulate transcription, processing and degradation of their sense cognate. For example, AtCOOLAIR, a cold-induced NAT, has been associated with transcriptional silencing of its cognate AtFLOWERING LOCUS C, but the importance of this as a trigger for vernalization is still in debate. Using tiling arrays, it was found that 24% of regulated protein coding genes were controlled by the circadian clock, while 7% of the protein coding genes have circadian-regulated NATs in Arabidopsis. Here we show that commercial sugarcane varieties have robust circadian rhythms driven by a central oscillator that is similar, but not identical to the Arabidopsis circadian clock. We also show that the proportion of probes that had rhythmic time courses s higher than the ones found in other plants and that the transcript levels in both sense and antisense directions are regulated by the circadian clock. We also show that a high proportion of probes associated with the harvesting and storage of energy from light and probes associated with DNA, RNA and protein synthesis are regulated by the clock circadian but the former are expressed during the light phase of the day and the latter are expressed during the dark phase. Taken together, our data suggest that the circadian clock is highly active in commercial sugarcane varieties and may be important to its high productivity and sucrose accumulation. Our oligoarray was designed based on the sugarcane expressed sequence tag database. The SUCEST database has 43,141 putative transcripts known as sugarcane assembled sequences. Every probe that passed quality controls and was unique to one SAS was used, minimizing any selection bias. However, there are several SAS within SUCEST that may correspond to the same coding gene, either because they represent different alleles, different paralogs, or because they are fragments of the same gene that were not assembled together. Furthermore, each probe was not designed to differentiate among alleles or duplications of the same gene. Thus, it was possible that the large proportions of circadian-regulated probes that we had identified did not correspond to a higher proportion of circadianregulated transcripts. In order to address this issue, we have selected 9 enzyme models associated with sucrose metabolism and compared their time courses among the different datasets. Eight of the sugarcane enzyme models had at least one rhythmic probe.
Viral miRNAs have potentially evolved to provide ideal tools for viruses to modulate both viral and cellular gene expression
This problem also calls for the development of highly efficient laboratory models of HPV infection. Altogether, five of the putative papillomavirus microRNAs were encoded by HPV 16, one by HPV 38, one by HPV 68, one by HPV 45 and one by HPV 6. HPV 68 belong to alpha-papillomaviruses, whereas HPV 38 is a beta-papillomavirus. None of the candidate HPV encoded microRNAs had similarity to known human microRNA sequences. Of the validated microRNAs, HPV16-miR-H1-1 is located within the E1 region of the coding strand, and HPV16-miR-H2-1 in the negative strand corresponding to the LCR region. Intriguingly, the HPV16-miRH2-1 sequence is present in a number of HPV 16 isolates but has a one-nucleotide deletion in the prototype sequence. Many of the isolates have been cloned from carcinoma tissues, suggesting that the ability to express this particular microRNA might promote carcinogenesis. Interestingly, the Orbifloxacin pre-microRNA sequence of HPV38-miR-H1, encoded by the E7 region, is shared by HPV types 22, 23, 120, 104 and 115, which are all members of the betapapillomavirus genus. Moreover, the pre-miRNA sequence of HPV45-miR-H1, encoded by the L1 region, is partially similar to HPV 16, suggesting Mepiroxol evolutionary divergence of viral miRNA function between HPV types. Although the deep sequencing read counts for the HPV 6 encoded miRNA were high, we were not able to validate it by qPCR, possibly due to the specific design of TaqMan assays. Because of the very short length of the miRNA there are very limited possibilities to alter the assay design if no results are obtained with the qPCR test. Several of the putative miRNA sequences were encoded by the negative DNA strand, which disagrees with the consensus that all papillomavirus transcripts originate from the positive strand of papillomavirus genomes. Although in this first report we did not study the mechanisms of transcription of HPV microRNAs, our methodology should practically exclude RNA degradation products or DNA from sequencing libraries. Viral miRNAs may also possess features that do not follow the canonical properties of human miRNAs. The precursor sequence of HPV16-miR-H1 is still uncertain and needs further validation of length and exact sequence. Due to low expression level of this miRNA we were not able to establish its exact length. Merkel cell polyomavirus encoded MCV-miR-M1-5p, which was first predicted from VMir and validated, and further identified by Illumina sequencing and validated by qRT-PCR, has a 59end 2-nt shift from the VMir predicted MCV-mir-M1 mature sequence, which has also been shown to exist and  be functional in vitro. Further studies are needed to prove whether the isomiRs presented here could also exist and be functional under some conditions. Entire tissue samples consisting of both healthy and infected cells were used for these studies. Robust signals were seen in cervical tissue in in situ hybridization, often colocalizing with and restricted to regions staining for p16INK4a, which is considered a surrogate marker for high-risk HPV oncogene activity. In situ hybridization also showed altered distribution of human miR-205, whose high expression has been reported before in CaSki cells and in cervical cancer tissues. miR-205 was also recently shown to promote proliferation of human cervical cancer cells. Although some viral miRNAs are occasionally expressed at high levels, low level of expression has also been shown biologically relevant, for example for Merkel cell polyomavirus miRNA. Those authors speculated that even low levels of viral miRNA expression might be sufficient to regulate host immune response. However, the signals in the in situ assays for the U6, miR-205 and HPV miRNAs cannot be directly compared as a measure of the expression level.
be functional in vitro. Further studies are needed to prove whether the isomiRs presented here could also exist and be functional under some conditions. Entire tissue samples consisting of both healthy and infected cells were used for these studies. Robust signals were seen in cervical tissue in in situ hybridization, often colocalizing with and restricted to regions staining for p16INK4a, which is considered a surrogate marker for high-risk HPV oncogene activity. In situ hybridization also showed altered distribution of human miR-205, whose high expression has been reported before in CaSki cells and in cervical cancer tissues. miR-205 was also recently shown to promote proliferation of human cervical cancer cells. Although some viral miRNAs are occasionally expressed at high levels, low level of expression has also been shown biologically relevant, for example for Merkel cell polyomavirus miRNA. Those authors speculated that even low levels of viral miRNA expression might be sufficient to regulate host immune response. However, the signals in the in situ assays for the U6, miR-205 and HPV miRNAs cannot be directly compared as a measure of the expression level.
Transfection of constitutive nuclear active FGFR1 also promotes neurite regeneration
Among the genes involved in neuronal differentiation, only a few have been studied in relation to Folinic acid calcium salt pentahydrate regulatory control by nuclear FGFR1, Nurs and RA receptors. Nuclear FGFR1 increases the expression of th, neurofilament, neuronal enolase and fgf-2 and chromatin immunoprecipitation experiments showed nuclear FGFR1, together with CBP and other DNA binding proteins, associates within the LOUREIRIN-B promoters of the th and fgf-2 genes. Yeast two-hybrid and coimmunopreciptation assays revealed that the FGFR1 tyrosine kinase domain binds directly to RSK1 Nterminal kinase. RSK1 binding promotes FGFR1 release from pre-Golgi to cytosol, increases the mobile population cytosolic of FGFR1 and facilitates nuclear accumulation of FGFR1. In the cell nucleus interaction of FGFR1 with RSK1 restricts the FGFR1 intra nuclear mobility and promotes RSK1 activation of CREB. In addition our recent studies showed that FGFR1 forms nuclear complexes with both the Nurr1 and Nur77 proteins. Given that RSK and Nur77 are fundamentally involved in nuclear signaling through both NGF and INFS, the possibility that NGF may utilize the INFS mechanism for neurodevelopmental and gene-activating functions has now been examined. We report that NGF promotes FGFR1 cytoplasmic-nuclear trafficking, in part, by inhibiting FGFR1 nuclear export. Furthermore, nuclear FGFR1 is essential for NGF-induced differentiation and transcriptional programming ofPC12 cells.FGFR1 binds to Nurtargeted regions of NGF-activated genes and augments NGF activation of ligand-independent function of Nur77/Nurr1. The present study provides a new perspective on the diverse actions of NGF which requires the neurodevelopmental INFS mechanism. Earlier studies from several laboratories  have provided in depth characterization of NGF-induced PC12 neuronal-like differentiation i.e., an outgrowth of neurites with growth cone-like endings accompanied by an up-regulation of the neurotransmitter biosynthetic enzyme, TH, neuronal b-III Tubulin, MAP-2, Neurofilament L, NMDAR1 protein and nicotinic acetylcholine receptor currents. Other studies have also indicated that NGF can evoke neuron-specific voltagedependent K+ and Na+ currents. In the present work we find that NGF-induced nuclear accumulation of FGFR1 is accompanied by exit from the cell cycle, an acquisition of neuronal morphology and the activation of th-Luc and other neuronal genes. The outgrowth of PC12 neurites was analyzed by measuring the length of neuritic processes using an established assay in cells co-transfected with plasmid expressing marker EGFP protein. Treatment of PC12 cells with NGF produced typical neurite outgrowth. In a loss of function experiment we cotransfected PC12 cells with dominant negative mutants of FGFR1, which lack the tyrosine kinase domain, form non-functional dimers with the endogenous receptor and compete with wild type FGFR1 for its nuclear targets. FGFR1 localizes to cytoplasmic membranes and cell nuclei. FGFR1, in which the signal peptide is replaced with a NLS, functions exclusively in the nucleus. Cells co-transfected with a control vector display short processes, however, when treated with NGF the processes elongate. The dominant negative receptors have no significant effect on neurite length in nonstimulated PC12 cells compared to controls. In contrast, cells transfected with FGFR1 or FGFR1 fail to extend neurites in response to NGF. In a gain of function experiment, PC12 co-transfected with full length constitutiveFGFR1, which contains a functional tyrosine kinase domain, display a marked 3-foldelongation of neurites indistinguishable from that induced by NGF. The effects of FGFR1 on neurite outgrowth are summarized in Fig. 3B. Additional experiments show that NGF increases the number of PC12 cells with elongated process while reducing the cell number with short processes. This effect is diminished by dominant negative FGFR1.
have provided in depth characterization of NGF-induced PC12 neuronal-like differentiation i.e., an outgrowth of neurites with growth cone-like endings accompanied by an up-regulation of the neurotransmitter biosynthetic enzyme, TH, neuronal b-III Tubulin, MAP-2, Neurofilament L, NMDAR1 protein and nicotinic acetylcholine receptor currents. Other studies have also indicated that NGF can evoke neuron-specific voltagedependent K+ and Na+ currents. In the present work we find that NGF-induced nuclear accumulation of FGFR1 is accompanied by exit from the cell cycle, an acquisition of neuronal morphology and the activation of th-Luc and other neuronal genes. The outgrowth of PC12 neurites was analyzed by measuring the length of neuritic processes using an established assay in cells co-transfected with plasmid expressing marker EGFP protein. Treatment of PC12 cells with NGF produced typical neurite outgrowth. In a loss of function experiment we cotransfected PC12 cells with dominant negative mutants of FGFR1, which lack the tyrosine kinase domain, form non-functional dimers with the endogenous receptor and compete with wild type FGFR1 for its nuclear targets. FGFR1 localizes to cytoplasmic membranes and cell nuclei. FGFR1, in which the signal peptide is replaced with a NLS, functions exclusively in the nucleus. Cells co-transfected with a control vector display short processes, however, when treated with NGF the processes elongate. The dominant negative receptors have no significant effect on neurite length in nonstimulated PC12 cells compared to controls. In contrast, cells transfected with FGFR1 or FGFR1 fail to extend neurites in response to NGF. In a gain of function experiment, PC12 co-transfected with full length constitutiveFGFR1, which contains a functional tyrosine kinase domain, display a marked 3-foldelongation of neurites indistinguishable from that induced by NGF. The effects of FGFR1 on neurite outgrowth are summarized in Fig. 3B. Additional experiments show that NGF increases the number of PC12 cells with elongated process while reducing the cell number with short processes. This effect is diminished by dominant negative FGFR1.
Exaggerated or dysfunctional cytokine response to acute influenza may actually contribute to severity
Their additional value may be in pinpointing areas of this response that may be amenable to dampening or other forms of therapeutic intervention. Thus, at the same time that one is treating the virus in at-risk individuals, it is conceivable that adjunctive therapy directed at abrogating or redirecting an overly exuberant host response could also be introduced to further improve prognosis. Tasocitinib 477600-75-2 Although this leap from the PI-103 research setting to the bedside still remains a challenge for the reasons cited, it is still reasonable to ask whether in the future even stronger prognostic utility or power might be found in analyzing select combinations or subsets of these markers, whether identified through statistical modeling or de novo as biologically plausible groupings. In addition to allowing for more intelligent utilization of available biomarkers, such an approach might further minimize justifiable concerns over potential over-interpretation of statistical associations seen in comparatively large batteries of single assay results not adjusted for multiple comparisons, such as those generated by the multiplex platforms in increasingly common use. In a limited look at this possibility, we found that combinations of multiple biomarkers from even four relatively indiscrete functional groupings did generate odds ratios for disease outcomes that were statistically robust, generalizable across both studies, and comparable to the most potent of the single biomarkers studied. The fact that elevated levels of certain biomarkers were found in each of the four groupings suggests a very broad and diverse immunologic response to acute influenza. But with further dissection of specific categories of markers it may be possible to delineate more precisely what role the disparate portions of the host response play in modulating the course of infection. Whether with  further exposition such a combination approach might prove to be either more powerful or, ideally, disease-specific than any of the single biomarkers highlighted remains yet to be determined. In any case, these preliminary results are intriguing and provide a rationale for performing more detailed analyses of these particular biomarkers as additional patients are accrued in these studies and additional progression events recorded. It is certainly possible, for example, that combinations of other biomarkers, or even different logical groupings of the same biomarkers into other categories based upon such factors as common cellular origin or relative positioning within the pro-inflammatory cascade, might offer results of equal or superior prognostic value or provide even greater insight into potentially deleterious aspects of the host immune response. The focus of this present analysis has been on the prognostic value of baseline biomarkers in predicting the subsequent course of disease specifically in Apdm09 virus-infected patients. Since both studies are ongoing on a multi-year basis in both the Northern and Southern Hemispheres, have been broadened to include patients presenting with all major subtypes of seasonal virus in circulation at the time, and now collect serial blood specimens for up to 60 days following enrollment, the intention is that these analyses can be expanded in at least two ways. One goal will be to map the kinetics in serum and plasma of each of the major biomarkers, singly and in groupings, from baseline.
further exposition such a combination approach might prove to be either more powerful or, ideally, disease-specific than any of the single biomarkers highlighted remains yet to be determined. In any case, these preliminary results are intriguing and provide a rationale for performing more detailed analyses of these particular biomarkers as additional patients are accrued in these studies and additional progression events recorded. It is certainly possible, for example, that combinations of other biomarkers, or even different logical groupings of the same biomarkers into other categories based upon such factors as common cellular origin or relative positioning within the pro-inflammatory cascade, might offer results of equal or superior prognostic value or provide even greater insight into potentially deleterious aspects of the host immune response. The focus of this present analysis has been on the prognostic value of baseline biomarkers in predicting the subsequent course of disease specifically in Apdm09 virus-infected patients. Since both studies are ongoing on a multi-year basis in both the Northern and Southern Hemispheres, have been broadened to include patients presenting with all major subtypes of seasonal virus in circulation at the time, and now collect serial blood specimens for up to 60 days following enrollment, the intention is that these analyses can be expanded in at least two ways. One goal will be to map the kinetics in serum and plasma of each of the major biomarkers, singly and in groupings, from baseline.